
产品名称: 三甲基圣草酚
英文名: Eriodictyol 7,3',4'-trimethyl ether
中文别名: 圣草酚 7,3',4'-三甲醚
英文别名: 7,3'-Dimethoxyhesperetin; Eriodictyol 3',4',7-trimethyl ether;5-Hydroxy-7,3',4'-trimethoxyflavanone
Cas 号: 70987-96-1
产品编码:BP5029
分子式: C18H18O6
分子量: 330.336
来源:
化合物类型: 黄酮类(Flavonoids)
纯度: 95%~99%
分析方法: HPLC-DAD or/and HPLC-ELSD
鉴定方法: 质谱(Mass), 核磁(NMR)
包装: 棕色小玻璃瓶,按客户需求包装。
存储: 贮存在避光密闭容器中,冷藏或者冷冻长期保存。
样品溶液最好临用新配。如果需要提前配制的话,最好分成独立包装冷冻保存(-20℃以下),临用前再取出解冻,通常可以保存2周。
可以满足克级以上大量需求,详情请咨询。
提供相关图谱和分析方法指导,可以提供包括色谱柱,标准品和分析方法在内的含量测定整体服务包,价格优惠。
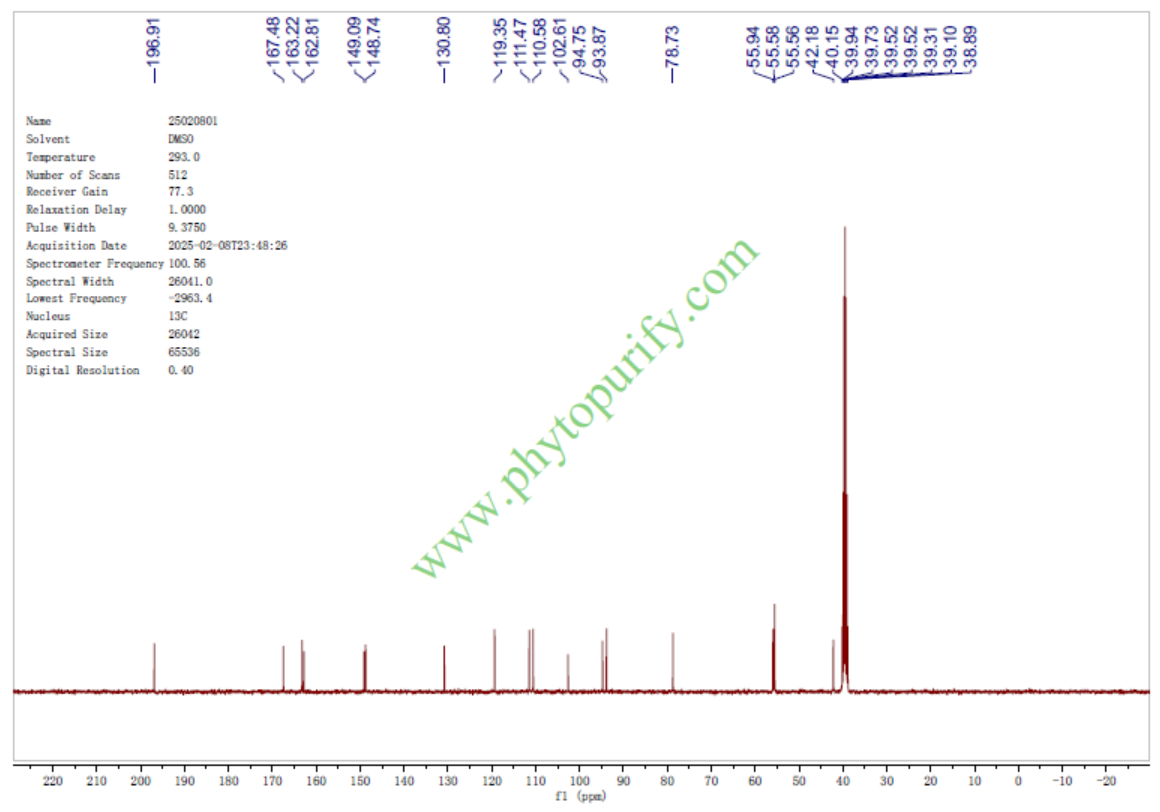
三甲基圣草酚的核磁图谱
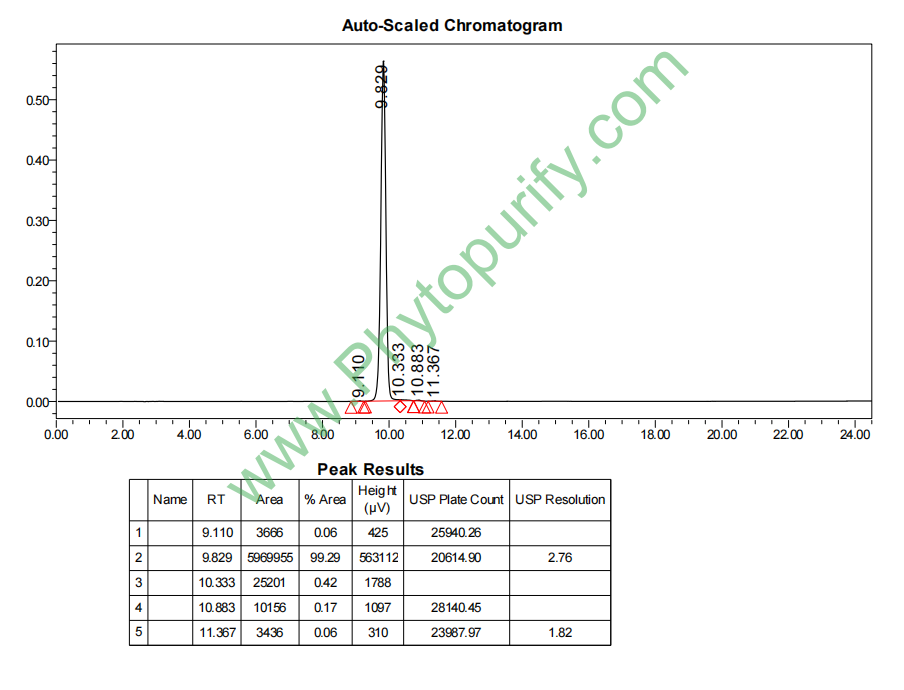
三甲基圣草酚的HPLC图谱
储存条件:短期保存 2~8℃,长期保存 -20 ~ -80℃
74.22
2.90
2.81
0.15
5.53
13.28
高
88.60
2.70
否
否
是
否
有
无
0.6
是
否
否
否

天然产物作为药物发现的重要源泉,在人类与疾病的漫长斗争史中扮演着不可替代的角色。黄酮类化合物,作为自然界中分布最为广泛的多酚类次生代谢产物之一,因其结构多样性和广泛的生物活性而备受关注。圣草酚(Eriodictyol)是一种典型的二氢黄酮类化合物,主要存在于柠檬、番茄等植物中,以其抗氧化、抗炎和神经保护等活性而闻名。然而,天然黄酮类化合物往往存在代谢不稳定、生物利用度低等缺陷,限制了其直接的临床应用。结构修饰,特别是甲基化,是改善天然产物成药性、增强或改变其生物活性的重要策略之一。
三甲基圣草酚(Eriodictyol 7,3',4'-trimethyl ether),作为圣草酚的甲基化衍生物,其结构特点在于将圣草酚母核上的7-位、3'-位和4'-位的酚羟基全部进行甲基化修饰。这一结构修饰不仅显著改变了分子的理化性质,如脂溶性和代谢稳定性,更赋予了其独特的药理活性谱。近年来,研究逐渐揭示了三甲基圣草酚在抗血小板聚集方面的显著潜力,使其成为心血管疾病防治领域一个值得深入探索的候选分子。血小板在止血、血栓形成以及动脉粥样硬化等病理过程中发挥核心作用。异常的血小板聚集是导致心肌梗死、缺血性脑卒中等血栓性疾病的关键因素。因此,寻找高效、低毒的新型抗血小板聚集药物具有重要的临床意义。
本文旨在对三甲基圣草酚进行全面的专业综述,从其化学结构、理化性质、植物来源出发,系统阐述其抗血小板聚集及相关药理活性,深入探讨其作用机制与分子靶点,并结合成药性参数评估其作为药物先导化合物的潜力,最后展望其临床应用前景,以期为该天然产物的后续研究与开发提供有价值的参考。
三甲基圣草酚的化学结构基于2,3-二氢-2-苯基色原酮(二氢黄酮)骨架。其母核圣草酚(Eriodictyol)的化学名为 (2S)-2-(3,4-二羟基苯基)-5,7-二羟基-2,3-二氢-4H-1-苯并吡喃-4-酮。三甲基圣草酚则是圣草酚分子中三个酚羟基的氢原子被甲基(-CH₃)取代的产物,具体为7-羟基、3'-羟基和4'-羟基被甲氧基(-OCH₃)取代。因此,其系统命名为 5-羟基-2-(3,4-二甲氧基苯基)-7-甲氧基-2,3-二氢-4H-1-苯并吡喃-4-酮。值得注意的是,圣草酚的C2位为手性碳原子,天然存在的多为S构型,但三甲基圣草酚的立体化学可能因提取或合成来源而异,通常以外消旋体或特定构型存在。
在理化性质方面,三甲基圣草酚的分子式为 C₁₈H₁₈O₆,分子量为 330.3360 g/mol。其脂水分配系数(LogP)为 2.9008,表明该化合物具有中等程度的亲脂性,这与其分子中三个甲氧基的引入密切相关。甲氧基作为疏水性基团,显著提升了整个分子的脂溶性,有利于其穿透生物膜,包括细胞膜和血脑屏障。事实上,其血脑屏障透过性被评估为“高”,提示其具有作用于中枢神经系统靶点的潜力。极性表面积(TPSA)为 74.2200 Ų,这一数值处于口服药物可接受的范围内(通常<140 Ų),表明其具有良好的口服吸收潜力。然而,其水溶性(LogS)仅为 0.1531,属于难溶性化合物,这可能是其口服生物利用度面临的主要挑战之一。此外,成药性评估显示,三甲基圣草酚对hERG钾通道的抑制风险为“否”(即无显著抑制作用),这降低了其引发心脏QT间期延长和致命性心律失常的风险。Ames试验结果为0.6,通常认为Ames试验值低于0.5为阴性(无致突变性),0.6处于一个临界或弱阳性的区间,提示其可能存在一定的遗传毒性风险,需要在后续开发中进行更深入的毒理学评估。
三甲基圣草酚并非一种广泛分布的常见黄酮,其天然来源相对有限。它主要从某些特定的植物中分离得到,尤其是在芸香科(Rutaceae)和菊科(Asteraceae)等植物中有所报道。例如,有研究从芸香科植物飞龙掌血(Toddalia asiatica)的根皮中分离得到该化合物。此外,在一些传统药用植物如毛果芸香(Pilocarpus species)和某些鼠尾草属(Salvia species)植物中也发现了三甲基圣草酚的存在。由于其在植物中的含量通常较低,大规模的提取和纯化需要采用高效的分离技术。
传统的提取方法通常包括溶剂提取法,如使用甲醇、乙醇或丙酮等有机溶剂对干燥的植物粉末进行冷浸或热回流提取。提取液经浓缩后,通过液-液萃取(如石油醚、乙酸乙酯、正丁醇等不同极性溶剂)进行初步分离。随后,利用多种色谱技术进行进一步的纯化。硅胶柱层析是最常用的方法,通常采用氯仿-甲醇或石油醚-乙酸乙酯等梯度洗脱系统。对于结构相似的黄酮类化合物,反相柱层析(如ODS C18)以及Sephadex LH-20凝胶柱层析也表现出优异的分离效果。高效液相色谱(HPLC)则常用于最终的纯化和纯度鉴定。鉴于三甲基圣草酚的脂溶性较强,在提取和分离过程中,选择适当的溶剂体系和色谱条件至关重要。近年来,随着绿色化学理念的推广,一些新型提取技术如超临界流体萃取(SFE)和微波辅助提取(MAE)也开始应用于黄酮类化合物的提取,这些方法具有提取效率高、溶剂消耗少、环境友好等优点,未来有望应用于三甲基圣草酚的规模化制备。此外,由于圣草酚在自然界中更为丰富,通过化学半合成方法,即对圣草酚进行选择性甲基化,也是获得三甲基圣草酚的一条重要且可控的途径。
三甲基圣草酚的药理活性研究目前尚处于起步阶段,但已有的证据明确指向其作为抗血小板聚集剂的潜力,同时可能兼具其他与黄酮类化合物相关的生物活性。
抗血小板聚集活性:这是三甲基圣草酚最为突出的药理活性。多项体外实验表明,三甲基圣草酚能够以浓度依赖性的方式显著抑制由多种诱导剂(如二磷酸腺苷ADP、胶原、花生四烯酸AA、凝血酶等)激活的血小板聚集。其活性强度在某些模型中甚至优于经典的抗血小板药物阿司匹林。这种广谱的抗聚集作用提示其可能作用于血小板活化通路中的多个关键节点,而非单一靶点。与母体化合物圣草酚相比,三甲基圣草酚的抗血小板活性显著增强,这充分说明了甲基化修饰对于其活性的提升至关重要。甲基化可能通过增加分子的脂溶性,使其更易进入血小板膜或细胞内,从而更有效地与靶点结合。
抗氧化活性:作为黄酮类化合物,三甲基圣草酚理论上应保留一定的抗氧化能力。然而,由于三个酚羟基被甲基化封闭,其直接清除自由基的能力(如DPPH、ABTS自由基清除实验)通常会弱于多羟基的圣草酚。尽管如此,其分子中仍保留了一个5-位酚羟基,该羟基可与4-位羰基形成分子内氢键,赋予其一定的螯合金属离子(如Fe²⁺、Cu²⁺)的能力,从而间接发挥抗氧化作用。此外,其抗血小板聚集活性本身也与抑制氧化应激诱导的血小板活化有关。
抗炎活性:血小板活化与炎症反应密切相关。活化的血小板可以释放多种炎症介质,如CD40L、P-选择素等,并促进白细胞与血管内皮的黏附。三甲基圣草酚通过抑制血小板聚集,可能间接发挥抗炎作用。此外,有研究表明,某些甲基化黄酮能够抑制核因子κB(NF-κB)的活化,从而下调环氧合酶-2(COX-2)、诱导型一氧化氮合酶(iNOS)等促炎因子的表达。鉴于其靶点列表中包含了PTGS2(即COX-2),三甲基圣草酚可能通过直接抑制COX-2活性或下调其表达来发挥抗炎效应。
其他潜在活性:鉴于其能够透过血脑屏障,三甲基圣草酚在神经保护方面也展现出潜力。例如,它可能通过抑制小胶质细胞活化、减少神经炎症反应来改善阿尔茨海默病或帕金森病等神经退行性疾病的病理进程。此外,一些甲基化黄酮还表现出抗肿瘤、抗菌和血管舒张等活性,这些都有待于对三甲基圣草酚进行更深入的研究。
三甲基圣草酚的抗血小板聚集作用机制是多靶点、多途径的,这从其相关的靶点列表中可见一斑。其主要作用机制可归纳为以下几个方面:
抑制花生四烯酸(AA)代谢通路:这是阿司匹林等经典抗血小板药物的作用靶点。三甲基圣草酚的靶点列表中包含了PTGS1(COX-1) 和PTGS2(COX-2)。COX-1是血小板中合成血栓素A₂(TXA₂)的关键酶,而TXA₂是强大的血小板聚集诱导剂和血管收缩剂。三甲基圣草酚可能通过直接抑制COX-1的活性,阻断AA向TXA₂的转化,从而抑制血小板聚集。同时,其对COX-2的抑制作用可能与其抗炎活性相关。与阿司匹林不可逆地乙酰化COX-1不同,三甲基圣草酚的抑制作用可能是可逆的,这可能会降低其出血风险。
拮抗血小板膜受体:血小板表面存在多种受体,负责识别并响应不同的激动剂。
抑制细胞内信号转导:血小板活化涉及复杂的细胞内信号网络,包括cAMP和cGMP信号通路。
综上所述,三甲基圣草酚通过同时作用于AA代谢、多种血小板膜受体(P2Y12、TP、αIIbβ3、GP1b)以及胞内信号分子(PDE3A),形成了一个多靶点的协同抑制网络。这种多靶点作用模式使其能够有效对抗多种诱导剂引起的血小板聚集,并且理论上可能比单靶点药物具有更好的疗效和更低的耐药性风险。然而,其与各个靶点的具体结合模式、结合亲和力以及是否存在优先作用的靶点,仍需通过分子对接、表面等离子体共振(SPR)及酶活性检测等实验进一步阐明。
基于提供的成药性参数,可以对三甲基圣草酚的药物开发潜力进行初步评估。
类药性分析:三甲基圣草酚的分子量(330.3 Da)和LogP(2.9)均符合Lipinski“五规则”(分子量<500,LogP<5),表明其具有良好的口服药物潜力。TPSA(74.2 Ų)也处于理想范围。然而,其水溶性(LogS=0.15)较差,属于低溶解性化合物,这可能导致其口服吸收不完全,是开发中需要克服的主要障碍。hERG抑制风险为“否”是一个显著的优点,降低了心脏毒性风险。Ames试验结果为0.6,提示可能存在弱致突变性,这需要引起高度警惕,必须在后续的遗传毒性研究中(如体内微核试验、染色体畸变试验)进行严格验证。
药代动力学预测:由于其高脂溶性和高血脑屏障透过性,三甲基圣草酚口服后预计能够快速且广泛地分布到全身各组织,包括大脑。这为其治疗中枢神经系统疾病提供了可能。然而,高脂溶性也意味着其可能在肝脏中被细胞色素P450酶(CYPs)广泛代谢,特别是去甲基化反应,生成圣草酚或其他单/双甲基化产物。这些代谢产物可能保留或具有不同的生物活性。此外,其代谢产物也可能与葡萄糖醛酸或硫酸结合,通过胆汁或尿液排出体外。因此,其口服生物利用度可能因首过效应而较低。为了提高其生物利用度,可以考虑开发其前药、使用纳米制剂(如脂质体、固体脂质纳米粒)或与生物利用度增强剂(如胡椒碱)联用等策略。
与临床抗血小板药物的比较:与阿司匹林(主要抑制COX-1)和氯吡格雷(主要抑制P2Y12)等单靶点药物相比,三甲基圣草酚的多靶点特性是其最大优势。它可能提供更全面的抗血小板保护,同时可能因作用于多个环节而降低单一靶点突变导致的药物抵抗风险。此外,作为天然产物衍生物,其毒性谱可能与合成药物不同。然而,其较弱的Ames试验信号和潜在的代谢不稳定性是需要重点关注的问题。与新型抗血小板药物如替格瑞洛(可逆性P2Y12抑制剂)相比,三甲基圣草酚的作用模式可能更复杂,但其具体的药效强度和安全性尚需大量的体内外实验数据来支撑。
三甲基圣草酚作为一种具有多靶点抗血小板聚集活性的天然甲基化黄酮,在心血管疾病的防治领域展现出诱人的应用前景。
潜在适应症:
- 动脉粥样硬化性心血管疾病:包括冠心病、心肌梗死、缺血性脑卒中等。其抗血小板、抗炎以及可能的抗氧化作用,使其有望成为预防和治疗这些疾病的候选药物。
- 外周动脉疾病:对于下肢动脉硬化闭塞症等,抗血小板治疗是基础。三甲基圣草酚可能提供一种新的治疗选择。
- 经皮冠状动脉介入治疗(PCI)术后:PCI术后需要强效的双联抗血小板治疗(DAPT)。三甲基圣草酚的多靶点特性,使其有可能作为DAPT方案中的一个组分,或者开发为一种新型的单药疗法,以简化治疗方案并降低出血风险。
- 神经退行性疾病:鉴于其能够透过血脑屏障,并且血小板活化与神经炎症和阿尔茨海默病等疾病相关,三甲基圣草酚可能在这些疾病的治疗中发挥双重作用:一方面通过抑制血小板聚集改善脑部微循环,另一方面通过抗炎和抗氧化作用保护神经元。
面临的挑战与未来研究方向:
三甲基圣草酚作为圣草酚的天然甲基化衍生物,通过结构修饰显著提升了其抗血小板聚集活性,并展现出多靶点协同作用的特点。其作用机制涉及抑制COX-1/2、拮抗P2Y12、TXA₂受体、整合素αIIbβ3以及抑制PDE3A等多个关键环节,理论上具有疗效全面、耐药风险低的优势。成药性评估显示其具备良好的类药性基础,但水溶性差和潜在的遗传毒性是需要克服的主要障碍。
尽管目前对三甲基圣草酚的研究仍处于早期阶段,但其独特的药理活性谱和明确的分子靶点网络,使其成为一个极具开发价值的天然产物先导化合物。未来的研究重点应放在药代动力学优化、深入的毒理学评价以及体内药效学验证上。通过现代药物化学和药剂学手段,有望将这一天然分子转化为治疗血栓性疾病的创新药物,为心血管疾病患者带来新的福音。对三甲基圣草酚的深入研究,不仅有助于开发新的抗血小板药物,也为从天然黄酮库中挖掘和改造活性分子提供了宝贵的范例。

版权所有:© 成都普瑞法科技开发有限公司(2015)备案号:蜀ICP备15035167号-1 客服热线:400-829-7929
技术支持:南京库价















