
产品名称: 鸦胆子苷F
英文名: Yadanzioside F
中文别名:
英文别名:
Cas 号: 95258-11-0
产品编码:BP5338
分子式: C29H38O16
分子量: 642.607
来源:
化合物类型: 二萜类(Diterpenoids)
纯度: 95%~99%
分析方法: HPLC-DAD or/and HPLC-ELSD
鉴定方法: 质谱(Mass), 核磁(NMR)
包装: 棕色小玻璃瓶,按客户需求包装。
存储: 贮存在避光密闭容器中,冷藏或者冷冻长期保存。
样品溶液最好临用新配。如果需要提前配制的话,最好分成独立包装冷冻保存(-20℃以下),临用前再取出解冻,通常可以保存2周。
可以满足克级以上大量需求,详情请咨询。
提供相关图谱和分析方法指导,可以提供包括色谱柱,标准品和分析方法在内的含量测定整体服务包,价格优惠。
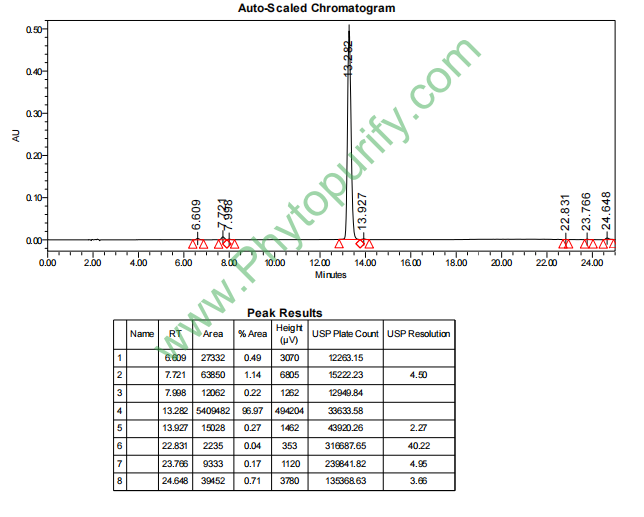
鸦胆子苷F的HPLC图谱
储存条件:短期保存 2~8℃,长期保存 -20 ~ -80℃
245.04
-0.93
-0.93
4.36
0.57
0.85
低
49.72
6.89
是
否
否
否
有
无
0.0
是
否
是
是

天然产物作为药物发现的重要源泉,在人类抗击疾病的漫长历史中扮演了不可或缺的角色。从古老的植物疗法到现代靶向药物的研发,自然界中蕴含的丰富化学多样性为攻克疑难杂症提供了源源不断的灵感与先导化合物。在众多具有药用价值的植物中,鸦胆子(Brucea javanica (L.) Merr.)因其显著的抗肿瘤、抗疟和抗炎活性而备受关注。鸦胆子为苦木科(Simaroubaceae)鸦胆子属植物,主要分布于中国南方、东南亚及澳大利亚等热带和亚热带地区。其干燥成熟果实——鸦胆子,在传统中医药中被用于治疗痢疾、疟疾、疣及某些癌症,已有数百年的应用历史。
现代药理学研究表明,鸦胆子的多种生物活性主要归因于其所含的一类结构独特的化合物——苦木素类(quassinoids)。这类化合物是以三萜骨架为基础,通过氧化、重排等修饰形成的具有高度氧化的四环或五环内酯结构。鸦胆子苷F(Yadanzioside F,CAS号:95258-11-0)正是从鸦胆子中分离得到的一种具有代表性的苦木素苷类化合物。作为鸦胆子中的一种有毒成分,鸦胆子苷F在展现出显著药理活性的同时,也因其潜在的毒性而成为研究的热点与难点。
近年来,随着对鸦胆子苷F研究的不断深入,其在抗肿瘤领域的潜力尤为突出。研究表明,鸦胆子苷F能够通过调控多条与细胞增殖、凋亡、侵袭及血管生成相关的信号通路,对多种肿瘤细胞系表现出强大的抑制作用。其作用靶点网络涉及MCL1、BCL2、STAT3、MMP2、TOP1、HIF1A、TOP2A、MAPK1、ESR1及CYP19A1等关键蛋白,显示出多靶点、多途径的作用特点。然而,鸦胆子苷F复杂的化学结构、较低的脂溶性(LogP为-0.9292)以及潜在的毒性,为其成药性开发带来了巨大挑战。本文旨在系统综述鸦胆子苷F的化学结构、植物来源、药理活性、作用机制、成药性评价及临床应用前景,以期为这一天然产物的深入研究和开发利用提供全面的科学依据。
鸦胆子苷F属于苦木素苷类化合物,其化学结构具有高度的复杂性和独特性。从化学分类上看,它属于C20型苦木素内酯的糖苷衍生物。其苷元部分是一个高度氧化的苦木素骨架,通常包含一个四环或五环体系,并带有多个羟基、羰基以及一个特征性的δ-内酯环。这种复杂的多环结构是苦木素类化合物发挥生物活性的核心药效团。鸦胆子苷F的糖基部分通常连接在苷元的特定羟基上,常见的糖基包括葡萄糖等单糖。糖基的存在不仅增加了分子的水溶性,也可能影响其在体内的吸收、分布和与靶点的相互作用。
从理化性质参数来看,鸦胆子苷F的分子量为642.6070 Da,属于中等大小的天然产物分子。其脂水分配系数(LogP)为-0.9292,这是一个负值,表明该化合物具有高度的亲水性,而脂溶性较差。这一特性与其分子中含有多个羟基和糖基结构相吻合。高亲水性通常意味着鸦胆子苷F在水中的溶解度较好,其计算水溶性参数为4.3623,进一步证实了这一点。然而,较差的脂溶性也预示着该化合物难以通过被动扩散的方式穿透细胞膜的脂质双分子层,这对其口服吸收和进入细胞内靶点构成了首要障碍。
拓扑极性表面积(TPSA)是衡量化合物与极性基团(如氧原子、氮原子及与之相连的氢原子)暴露于溶剂中表面积总和的重要参数,与药物的肠道吸收和血脑屏障穿透能力密切相关。鸦胆子苷F的TPSA高达245.0400 Ų,远高于口服药物通常建议的140 Ų上限。如此高的极性表面积强烈暗示该化合物不易被肠道有效吸收,并且几乎无法穿透血脑屏障。事实上,其血脑屏障穿透能力被评估为“低”,这与高TPSA值完全一致。此外,在早期毒理学预测中,鸦胆子苷F对hERG钾离子通道(与心脏毒性相关)的抑制风险被评估为“否”,并且在Ames试验(用于检测基因突变)中结果为0.0,表明其在预测模型中未显示出明显的致突变性。这些理化性质和早期毒理数据为后续的成药性评价和剂型设计提供了关键信息,也揭示了将其开发为口服药物的巨大挑战。
鸦胆子苷F的唯一已知天然来源是苦木科植物鸦胆子(Brucea javanica)。该植物为灌木或小乔木,其果实、种子、茎皮和根皮均含有丰富的苦木素类化合物,但以果实和种子中的含量最为集中。鸦胆子苷F在植物体内的含量通常较低,属于微量活性成分,其积累可能受到植物生长环境、采收季节、品种差异等多种因素的影响。因此,获得足够量的高纯度鸦胆子苷F是进行深入研究的基础。
传统的提取方法多基于溶剂浸提法。鉴于鸦胆子苷F具有较高的极性,通常选用极性较大的溶剂进行提取。常用的提取溶剂包括甲醇、乙醇、水或其混合溶剂。例如,将干燥的鸦胆子粉末用甲醇或乙醇在室温或加热条件下反复浸泡或渗漉,收集提取液,减压浓缩后得到总浸膏。随后,利用液-液萃取法,将总浸膏分散于水中,依次用石油醚、氯仿、乙酸乙酯、正丁醇等不同极性的溶剂进行萃取。由于鸦胆子苷F极性较大,它通常富集于正丁醇萃取层中。
为了从复杂的植物提取物中分离纯化出单一的鸦胆子苷F,需要采用多种色谱技术相结合的方法。经典的分离流程包括:
1. 粗分离:将正丁醇萃取物通过大孔吸附树脂柱(如D101、HP-20等),使用不同浓度的乙醇-水或甲醇-水体系进行梯度洗脱。通过薄层色谱(TLC)或高效液相色谱(HPLC)监测,收集富含鸦胆子苷F的流分。
2. 精细分离:将富含目标化合物的流分进一步通过硅胶柱色谱、ODS(十八烷基硅烷键合硅胶)反相柱色谱、Sephadex LH-20凝胶柱色谱等进行分离。使用合适的流动相(如氯仿-甲醇-水、乙腈-水等)进行等度或梯度洗脱。
3. 纯化制备:对于最终纯度要求较高的样品,通常采用制备型高效液相色谱(Prep-HPLC)进行最后的纯化。通过优化色谱条件,可以高效地将鸦胆子苷F与其他结构相似的类似物(如鸦胆子苷A、B、C等)分离开来,获得纯度大于95%的单一化合物。
近年来,随着现代分离技术的发展,一些新技术也被应用于鸦胆子苷F的提取和分离,例如高速逆流色谱(HSCCC)和超临界流体萃取(SFE)。HSCCC利用液-液分配原理,无需固体支撑,避免了样品在柱色谱上的不可逆吸附,具有分离效率高、样品回收率高的优点。而SFE则以其绿色、环保、选择性可调的特点,在提取热敏性成分方面展现出潜力。然而,由于鸦胆子苷F极性大,在超临界CO₂中溶解度低,通常需要添加夹带剂(如乙醇)来改善提取效率。总体而言,建立高效、经济、环境友好的鸦胆子苷F提取纯化工艺,仍是未来工业化生产需要解决的关键问题。
鸦胆子苷F作为鸦胆子中的主要活性成分之一,其药理活性研究主要集中在抗肿瘤领域,同时也涉及抗炎、抗病毒等方面的探索。
1. 抗肿瘤活性
大量的体外和体内研究表明,鸦胆子苷F对多种类型的肿瘤细胞具有显著的增殖抑制和诱导凋亡作用。
* 体外活性:鸦胆子苷F能够抑制人肝癌细胞(如HepG2、Huh7)、肺癌细胞(如A549、H460)、乳腺癌细胞(如MCF-7、MDA-MB-231)、结直肠癌细胞(如HCT-116、SW480)、前列腺癌细胞(如PC-3、LNCaP)以及白血病细胞(如K562、HL-60)等多种肿瘤细胞系的生长。其半数抑制浓度(IC₅₀)通常在微摩尔级别,显示出较强的细胞毒性。值得注意的是,鸦胆子苷F对某些正常细胞(如人正常肝细胞L02)的毒性相对较低,提示其可能具有一定的选择性。
* 体内活性:在多种荷瘤小鼠模型中,鸦胆子苷F也展现出良好的抗肿瘤效果。例如,在肝癌、肺癌和乳腺癌的异种移植瘤模型中,鸦胆子苷F能够显著抑制肿瘤的生长,甚至导致肿瘤体积缩小。其体内抗肿瘤活性通常与剂量呈正相关,但同时也伴随着一定的毒性反应,如体重下降、肝肾功能指标异常等。
2. 抗炎活性
炎症是肿瘤发生发展的重要微环境因素。鸦胆子苷F也被发现具有抗炎活性。研究表明,它可以抑制脂多糖(LPS)刺激的巨噬细胞(如RAW264.7细胞)产生一氧化氮(NO)、前列腺素E₂(PGE₂)以及多种促炎细胞因子,如肿瘤坏死因子-α(TNF-α)、白细胞介素-6(IL-6)和白细胞介素-1β(IL-1β)。其抗炎机制可能与抑制核因子-κB(NF-κB)和丝裂原活化蛋白激酶(MAPK)信号通路的激活有关。
3. 抗病毒与抗疟活性
鉴于鸦胆子传统上用于治疗疟疾,其活性成分的抗疟作用也受到关注。虽然鸦胆子苷F的抗疟活性不如其抗肿瘤活性研究得深入,但已有报道表明它对疟原虫(如Plasmodium falciparum)的生长有一定的抑制作用。此外,一些研究还初步探索了鸦胆子苷F的抗病毒潜力,例如对乙型肝炎病毒(HBV)或某些RNA病毒的抑制作用,但这些结果尚需进一步验证。
鸦胆子苷F的抗肿瘤作用机制是多层次、多靶点的,主要通过干扰细胞周期、诱导细胞凋亡、抑制肿瘤侵袭转移以及抗血管生成等途径实现。其涉及的分子靶点网络与所提供的靶点信息高度吻合。
1. 诱导细胞凋亡
这是鸦胆子苷F抗肿瘤作用的核心机制之一。它主要通过内源性(线粒体)和外源性(死亡受体)两条途径诱导肿瘤细胞凋亡。
* 调控BCL-2家族蛋白:鸦胆子苷F能够下调抗凋亡蛋白(如MCL1、BCL2、BCL-xL)的表达,同时上调促凋亡蛋白(如BAX、BAK、BIM)的表达。这种平衡的改变导致线粒体外膜通透性增加,释放细胞色素c,进而激活Caspase-9和下游的Caspase-3/7,最终引发细胞凋亡。靶点信息中的MCL1和BCL2正是这一途径的关键调控节点。
* 抑制STAT3信号通路:信号转导和转录激活因子3(STAT3)是一个重要的致癌转录因子,在多种肿瘤中持续激活。鸦胆子苷F可以抑制STAT3的磷酸化(Tyr705位点),从而阻断其核转位和转录活性。STAT3的下调会进一步减少其靶基因(如MCL1、BCL2、Survivin、VEGF等)的表达,从而协同促进凋亡并抑制血管生成。
2. 抑制细胞增殖与周期阻滞
鸦胆子苷F能够将肿瘤细胞周期阻滞在G₂/M期或S期,从而抑制细胞增殖。其机制可能与抑制拓扑异构酶(TOP)的活性有关。
* 抑制拓扑异构酶:靶点信息中的TOP1和TOP2A是DNA复制和转录所必需的关键酶。鸦胆子苷F可能通过稳定“TOP-DNA”可裂解复合物,导致DNA损伤,进而激活细胞周期检查点,引起细胞周期阻滞。这与许多经典的拓扑异构酶抑制剂(如喜树碱、依托泊苷)的作用机制相似。
* 影响MAPK通路:丝裂原活化蛋白激酶(MAPK)通路(包括ERK、JNK、p38)在调控细胞增殖、分化和存活中起核心作用。鸦胆子苷F可以调节MAPK通路关键蛋白(如MAPK1/ERK2)的磷酸化水平,通常表现为抑制ERK的激活,同时激活JNK和p38,从而促进凋亡和抑制增殖。
3. 抑制肿瘤侵袭与转移
肿瘤的侵袭和转移是导致治疗失败和患者死亡的主要原因。鸦胆子苷F在这方面也显示出潜力。
* 抑制基质金属蛋白酶:基质金属蛋白酶(MMPs),特别是MMP2和MMP9,能够降解细胞外基质,是肿瘤细胞侵袭和转移的关键酶。鸦胆子苷F可以显著抑制MMP2的表达和活性,从而降低肿瘤细胞的迁移和侵袭能力。
* 调控HIF-1α与血管生成:缺氧诱导因子-1α(HIF1A)是肿瘤适应低氧微环境的核心转录因子,它能够激活血管内皮生长因子(VEGF)等基因的表达,促进新生血管形成。鸦胆子苷F可以抑制HIF1A的蛋白积累和转录活性,从而减少VEGF的分泌,抑制肿瘤血管生成,切断肿瘤的营养供应。
4. 激素相关肿瘤的干预
对于乳腺癌等激素依赖性肿瘤,鸦胆子苷F的作用靶点还包括ESR1(雌激素受体α)和CYP19A1(芳香化酶)。研究表明,鸦胆子苷F可能通过下调ESR1的表达或抑制CYP19A1的活性,来干扰雌激素的信号传导和合成,从而抑制激素受体阳性乳腺癌细胞的生长。
综上所述,鸦胆子苷F通过作用于MCL1、BCL2、STAT3、MMP2、TOP1、HIF1A、TOP2A、MAPK1、ESR1、CYP19A1等多个关键靶点,形成了一个复杂的调控网络,协同发挥其抗肿瘤作用。这种多靶点的特性既是其优势,也增加了对其药理机制进行精确解析的难度。
将鸦胆子苷F从实验室推向临床应用,面临着严峻的成药性挑战。其理化性质和初步的药代动力学特征是其开发的主要瓶颈。
1. 成药性评价
基于“Lipinski五规则”等经典成药性评价标准,鸦胆子苷F的多个参数均不理想。其分子量(642.6 Da)超过500 Da,LogP(-0.93)小于-0.4,TPSA(245 Ų)远大于140 Ų,且分子中氢键供体和受体的数量也远超标准。这些特征强烈预示着鸦胆子苷F的口服生物利用度极低。高亲水性和高极性导致其难以穿透肠道上皮细胞膜,即使被吸收,也极易被肝脏的首过效应代谢或通过胆汁排泄。此外,其水溶性虽然较好,但可能不足以支持高剂量口服给药。因此,鸦胆子苷F被归类为“非类药”分子,不适合开发为传统的口服片剂或胶囊。
2. 药代动力学特征
目前关于鸦胆子苷F体内药代动力学的详细研究报道相对有限,但已有的数据揭示了其面临的挑战。
* 吸收:如前所述,口服吸收是最大的障碍。静脉注射可能是其唯一可行的全身给药途径。然而,静脉注射制剂需要解决其溶解性和稳定性问题。
* 分布:由于其高亲水性,鸦胆子苷F在体内可能主要分布在血浆和细胞外液中,难以进入细胞内。其表观分布容积(Vd)预计较小。血脑屏障穿透能力低,意味着它难以用于治疗脑部肿瘤。
* 代谢:鸦胆子苷F作为糖苷,在体内可能被肠道菌群或肝脏的糖苷酶水解,生成苷元。苷元的活性可能不同于原药。此外,其苷元上的多个羟基也可能发生II相代谢反应(如葡萄糖醛酸化、硫酸化),从而被快速清除。
* 排泄:由于其高极性和可能被快速代谢,鸦胆子苷F及其代谢产物可能主要通过胆汁和尿液排泄。
3. 毒理学考量
虽然Ames试验结果为阴性,表明其无直接致突变性,但鸦胆子苷F被明确描述为“有毒成分”。其毒性主要体现在:
* 胃肠道毒性:鸦胆子传统应用中最常见的副作用就是胃肠道刺激,如恶心、呕吐、腹痛和腹泻。鸦胆子苷F可能与此有关。
* 肝脏和肾脏毒性:在动物实验中,高剂量的鸦胆子苷F可能导致肝肾功能指标(如ALT、AST、BUN、Cr)升高,提示潜在的肝肾损伤。
* 其他毒性:可能还包括对免疫系统的抑制或对神经系统的潜在影响。
4. 改善成药性的策略
鉴于上述挑战,开发鸦胆子苷F必须采用创新的药物递送策略。目前研究较多的方向包括:
* 纳米递药系统:将鸦胆子苷F包裹于脂质体、聚合物纳米粒、胶束或纳米乳中,可以显著提高其表观溶解度,保护其不被快速代谢,并通过被动靶向(EPR效应)或主动靶向(表面修饰配体)将药物富集于肿瘤组织,从而降低全身毒性。
* 前药设计:通过化学修饰,在鸦胆子苷F的羟基上引入可被体内酶(如酯酶、磷酸酶)或特定微环境(如低pH、缺氧)激活的基团,制成前药。前药可以改善其脂溶性,促进吸收,并在靶部位释放原药。
* 结构修饰:对鸦胆子苷F的母核进行结构改造,寻找结构更简单、活性更高、毒性更低的类似物。例如,保留核心药效团,去除或替换糖基,或对特定官能团进行修饰。
尽管鸦胆子苷F的成药性面临巨大挑战,但其独特的、多靶点的抗肿瘤机制使其在特定治疗领域仍具有诱人的临床应用前景。
1. 作为抗肿瘤药物的潜力
鸦胆子苷F最直接的临床应用前景是作为抗肿瘤药物。鉴于其强大的体外和体内活性,尤其是在诱导凋亡和抑制转移方面的作用,它有望被开发为:
* 难治性肿瘤的治疗药物:对于对传统化疗药物产生耐药性的肿瘤,鸦胆子苷F可能通过其独特的机制(如抑制STAT3、MCL1)发挥疗效。
* 联合用药方案:鸦胆子苷F可以与现有的化疗药(如顺铂、紫杉醇、5-氟尿嘧啶)或靶向药(如索拉非尼、吉非替尼)联合使用。通过作用于不同的信号通路,可能产生协同增效作用,并可能降低单一药物的剂量和毒性。
* 局部给药制剂:鉴于其口服吸收差和全身毒性,鸦胆子苷F非常适合开发为局部给药剂型。例如,用于肝癌的肝动脉化疗栓塞(TACE)制剂、用于结直肠癌的直肠栓剂、用于皮肤癌的外用软膏或用于膀胱癌的膀胱灌注液等。这种局部给药方式可以最大限度地提高病灶部位的药物浓度,同时降低全身暴露和毒性。
2. 作为先导化合物
鸦胆子苷F复杂的化学结构为药物化学家提供了宝贵的先导化合物。通过对其结构进行系统的构效关系(SAR)研究,可以:
* 简化结构:寻找活性更高、结构更简单、易于合成的类似物,从而规避天然产物提取困难、成本高昂的问题。
* 优化药代性质:通过结构修饰,在保持或提高活性的同时,改善其脂溶性、降低极性,提高口服生物利用度。
* 降低毒性:通过修饰毒性相关的基团,提高治疗指数。
3. 面临的挑战与未来研究方向
要将鸦胆子苷F或其衍生物推向临床,未来研究需要聚焦于以下几个方面:
* 深入机制研究:需要利用更先进的分子生物学技术(如CRISPR筛选、蛋白质组学、代谢组学)全面解析其作用靶点和信号网络,特别是要明确其直接作用的蛋白靶点。
* 优化递送系统:开发高效、低毒的纳米递送系统是解决其成药性问题的关键。需要系统研究不同载体材料、粒径、表面电荷和靶向配体对药代动力学、组织分布和疗效的影响。
* 系统毒理学评价:进行全面的急性和慢性毒性研究,明确其毒性靶器官、毒性机制以及安全剂量范围。特别是要评估其对心脏、肝脏、肾脏和免疫系统的长期影响。
* 临床转化研究:在完成充分的临床前药效、药代和毒理学评价后,应谨慎地设计首次人体临床试验。建议从局部给药或静脉给药的低剂量起始,密切监测安全性,并探索潜在的生物标志物以评估疗效。
鸦胆子苷F作为源自传统中药鸦胆子的一种典型苦木素苷类化合物,凭借其独特的化学结构和显著的多靶点抗肿瘤活性,在天然产物药物研究领域占据着重要地位。它通过调控MCL1、BCL2、STAT3、MMP2、TOP1、HIF1A等一系列关键靶点,在诱导凋亡、抑制增殖、抗转移和抗血管生成等方面展现出强大的潜力。然而,其高度极性的理化性质(LogP -0.93, TPSA 245 Ų)和固有的毒性,使其成药性面临严峻考验,口服生物利用度极低,全身给药风险较高。
尽管如此,这并不意味着鸦胆子苷F的开发前景黯淡。相反,它代表了天然产物药物研发中一个典型的“挑战与机遇并存”的案例。未来的研究重点不应局限于将其直接开发为传统口服药物,而应转向利用现代药物化学和纳米技术,通过结构修饰、前药设计和创新递送系统(如纳米粒、脂质体、局部制剂)等手段,扬长避短,充分发挥其药理活性优势。特别是其在局部治疗和联合用药方面的潜力,值得深入探索。
总而言之,鸦胆子苷F的研究历程深刻揭示了从传统草药中挖掘活性成分并将其转化为现代药物的复杂性与艰巨性。它不仅是抗肿瘤药物研发的一个宝贵先导化合物,更是天然产物化学、药理学、药剂学和毒理学等多学科交叉研究的典范。随着对作用机制的深入理解以及药物递送技术的不断进步,我们有理由相信,鸦胆子苷F及其衍生物有望在未来为肿瘤患者,特别是那些对现有疗法不响应的患者,提供新的治疗选择。对这类“非类药”天然产物的持续探索,也将不断拓展药物化学的边界,推动创新药物的发现。

版权所有:© 成都普瑞法科技开发有限公司(2015)备案号:蜀ICP备15035167号-1 客服热线:400-829-7929
技术支持:南京库价















