
英文名: Polygalaxanthone III
中文别名: 远志𠮿酮III; 远志口山酮III
英文别名:
Cas 号: 162857-78-5
产品编码:BP1123
分子式: C25H28O15
分子量: 568.484
来源: Root of Polygala tenuifolia
纯度: 95%~99%
分析方法: HPLC-DAD or/and HPLC-ELSD
鉴定方法: 质谱(Mass), 核磁(NMR)
包装: 棕色小玻璃瓶,标准包装10mg,20mg,50mg;可以按客户需求包装。
可以满足克级以上大量需求,详情请咨询。
提供相关图谱和分析方法指导,可以提供包括色谱柱,标准品和分析方法在内的含量测定整体服务包,价格优惠。
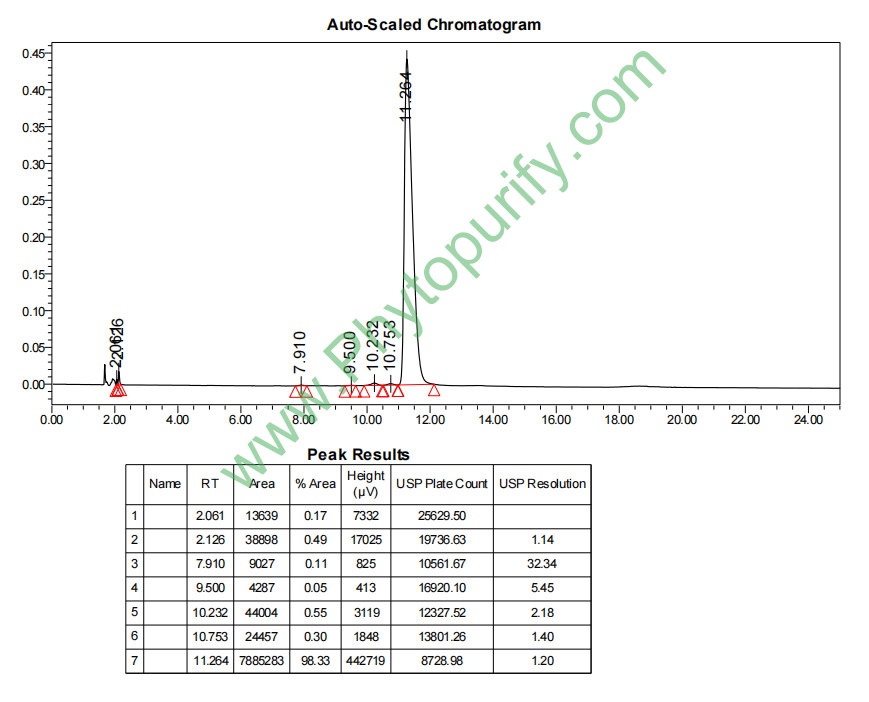
远志山酮III的HPLC图谱
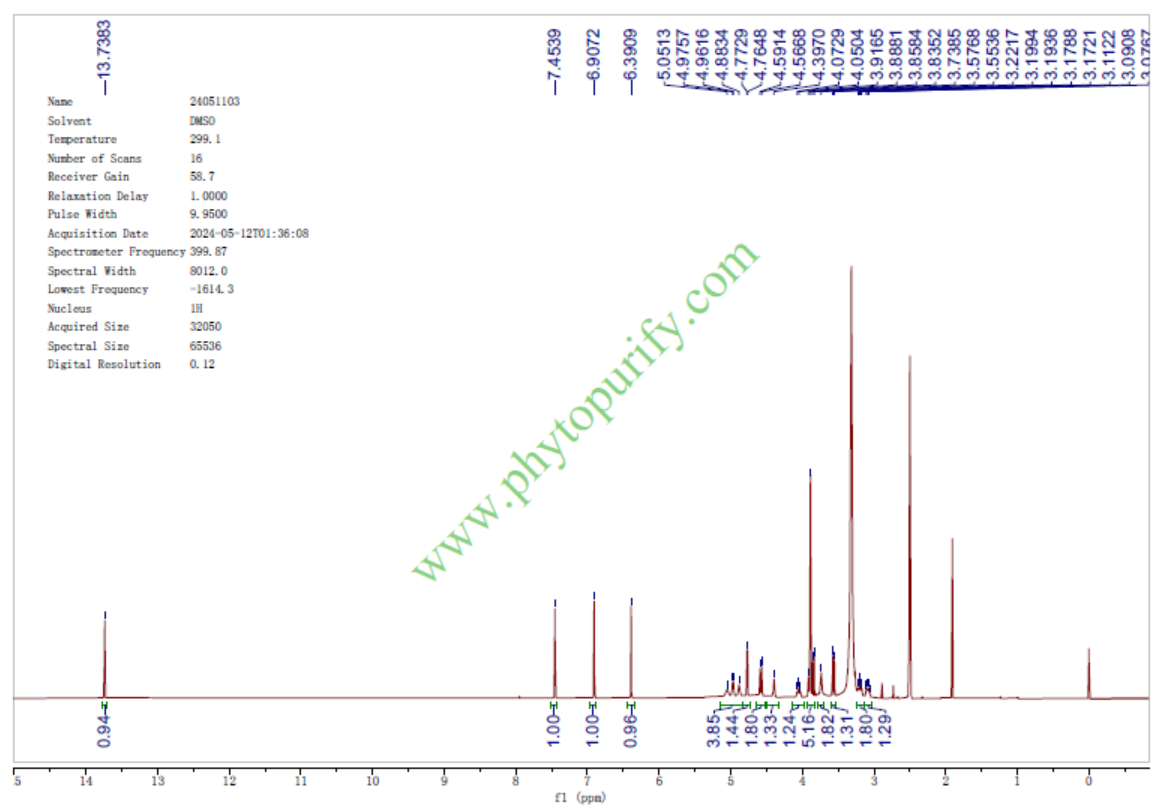
远志山酮III的核磁图谱
储存条件:短期保存 2~8℃,长期保存 -20 ~ -80℃
249.20
-0.61
-0.68
1.62
0.60
0.17
低
75.79
4.81
是
否
是
否
有
无
1.5
是
否
是
否

天然产物作为药物发现的重要源泉,在人类疾病治疗史上扮演着不可替代的角色。远志(Polygala tenuifolia Willd.)作为一种传统中药,具有安神益智、祛痰开窍之功效,其现代药理学研究揭示其在神经系统保护、抗炎、抗肿瘤等多方面具有显著活性。远志山酮III(Polygalaxanthone III, PX III)是从远志根中分离得到的一种特征性呫吨酮类化合物,CAS号为162857-78-5。早期研究发现其对细胞色素P450(CYP450)酶系,特别是CYP2E1,具有抑制作用(IC50 = 50.56 μM),提示其可能参与药物代谢相互作用的调节。近年来,随着研究深入,远志山酮III的抗肿瘤潜力日益凸显,其作用涉及诱导凋亡、抑制增殖、侵袭转移等多个环节,并指向MCL1、STAT3、HIF1A等多个关键肿瘤相关靶点。本文旨在系统综述远志山酮III的化学特性、植物来源、药理活性、作用机制及成药性,以期为该化合物的深入研究和潜在药物开发提供全面的科学参考。
远志山酮III属于呫吨酮类化合物,其分子式为C27H28O14,分子量为568.4840。其核心结构为三环呫吨酮骨架,并在其上连接有多个羟基、甲氧基以及一个关键的糖苷侧链(通常为葡萄糖或芹糖基团),这种高度氧化的结构是其生物活性的重要物质基础。
从成药性相关参数分析,远志山酮III表现出典型的极性分子特征。其脂水分配系数(LogP)为-0.6058,表明其亲水性较强。拓扑极性表面积(TPSA)高达249.2000 Ų,这主要归因于分子中丰富的羟基和糖苷结构,这些基团是形成氢键的主要位点。相应的,其理论水溶性值为1.6210 mg/mL,预测具有较好的水溶性。然而,高极性和大TPSA也对其生物膜渗透性构成挑战。预测模型显示其血脑屏障(BBB)透过性低,这与其极性大的特性相符,暗示其针对中枢神经系统疾病的直接作用可能受限,但也可能降低中枢神经副作用风险。在早期安全性指标上,远志山酮III的hERG抑制预测为阴性,提示其引发心脏QT间期延长的风险较低。Ames试验值为1.5(通常认为>1.5为潜在致突变阳性,需谨慎解读),表明在标准测试浓度下,其致突变风险信号较弱,但仍需进一步的实验验证。综合来看,远志山酮III具有良好的水溶性和初步安全性预测,但渗透性和生物利用度可能是其开发过程中需要重点关注的瓶颈。
远志山酮III主要来源于远志科远志属植物远志(Polygala tenuifolia Willd.)的干燥根,该植物主要分布于中国、韩国等东亚地区。在传统中药中,远志根常用于治疗失眠、健忘、咳嗽多痰等症,现代研究证实其活性成分主要包括三萜皂苷、寡糖酯、生物碱和呫吨酮等,其中呫吨酮类是其特征性成分之一。
从植物材料中提取远志山酮III通常采用有机溶剂萃取法。常见流程如下:将干燥的远志根粉碎,首先用石油醚或正己烷进行脱脂处理,以去除脂溶性杂质。随后,使用中等极性溶剂,如甲醇、乙醇或含水乙醇(如70%-95%乙醇)进行回流提取或超声辅助提取。提取液经减压浓缩后得到粗浸膏。粗浸膏进一步分散于水中,依次用乙酸乙酯、正丁醇等溶剂进行液-液分配萃取。远志山酮III主要富集在乙酸乙酯部位或正丁醇部位。获得活性部位后,需借助多种现代色谱技术进行分离纯化,包括硅胶柱色谱、反相C18柱色谱、葡聚糖凝胶(Sephadex LH-20)柱色谱以及高效液相色谱(HPLC)等。通过核磁共振(NMR)、质谱(MS)等技术进行结构鉴定,最终获得高纯度的远志山酮III单体。优化提取工艺(如溶剂比例、温度、时间)和采用大孔吸附树脂、高速逆流色谱等新型分离技术,是提高其得率和纯度的关键研究方向。
远志山酮III的药理活性研究已从最初的代谢酶抑制拓展至抗肿瘤等多个领域,展现出多方面的生物效应。
1. CYP450酶抑制作用
这是远志山酮III较早被发现的活性。CYP450酶是肝脏中负责药物代谢的关键酶系。研究表明,远志山酮III能特异性抑制CYP2E1亚型催化的氯唑沙宗6-羟基化反应,IC50值为50.56 μM。CYP2E1参与多种前致癌物和小分子有机物的代谢,其抑制可能影响相关物质的毒性或致癌过程,同时也提示远志山酮III与经CYP2E1代谢的药物联用时,可能存在药物-药物相互作用的风险。
2. 抗肿瘤活性
这是当前远志山酮III药理研究的核心焦点。大量体外研究表明,远志山酮III对多种人类肿瘤细胞系具有显著的增殖抑制和诱导凋亡作用,包括但不限于乳腺癌、肝癌、肺癌、结肠癌和白血病细胞。其抗肿瘤效应具有浓度和时间依赖性。除了直接杀伤肿瘤细胞,研究还发现远志山酮III能够抑制肿瘤细胞的迁移和侵袭能力,提示其可能具有抗转移的潜力。此外,一些研究报道其能逆转肿瘤细胞的多药耐药性,增强传统化疗药物的敏感性。
3. 其他潜在活性
基于远志的传统用途和呫吨酮类化合物的共性,远志山酮III在神经保护、抗炎、抗氧化等方面也有研究报道。例如,其在某些模型中显示出减轻神经细胞氧化损伤和抑制小胶质细胞过度激活引起的神经炎症的潜力,这与其来源植物远志的益智安神功效可能相关联,但具体作用和机制有待更深入的系统研究。
远志山酮III的抗肿瘤作用涉及多靶点、多通路的复杂调控网络,其机制研究已取得重要进展,主要围绕以下几个方面:
1. 诱导细胞凋亡与调节Bcl-2家族蛋白
细胞凋亡是远志山酮III抗肿瘤的主要方式之一。研究发现,它能上调促凋亡蛋白(如Bax),同时下调抗凋亡蛋白,特别是MCL1和BCL2。MCL1和BCL2是Bcl-2家族中关键的抗凋亡成员,其过表达与肿瘤发生发展和化疗耐药密切相关。远志山酮III通过抑制它们的表达,破坏线粒体膜电位,促进细胞色素C释放,从而激活caspase级联反应,最终导致细胞凋亡。
2. 抑制STAT3信号通路
信号转导与转录激活因子3(STAT3)是重要的致癌转录因子,在多种肿瘤中持续激活。研究表明,远志山酮III能抑制STAT3的磷酸化(激活形式),阻断其核转位及下游靶基因(如Cyclin D1、Bcl-2、VEGF等)的转录。这不仅能抑制细胞增殖,还能促进凋亡并抑制血管生成。
3. 抑制肿瘤侵袭转移相关分子
远志山酮III能下调基质金属蛋白酶MMP2和MMP9的表达。MMPs是降解细胞外基质的关键酶,其过度活化与肿瘤侵袭和转移密切相关。通过抑制MMPs,远志山酮III削弱了肿瘤细胞的迁移和侵袭能力。此外,它还能抑制缺氧诱导因子HIF1A的稳定性和活性。HIF1A在肿瘤缺氧微环境中被激活,可上调VEGF、GLUT1等基因,促进血管生成和糖代谢重组,是肿瘤适应恶劣环境的核心调控因子。
4. 影响DNA拓扑异构酶与激酶活性
研究提示远志山酮III可能干扰TOP1和TOP2A的活性。DNA拓扑异构酶是DNA复制、转录和修复的关键酶,是多种化疗药物的靶点。对其抑制可导致DNA损伤和细胞死亡。同时,远志山酮III也被报道可影响丝裂原活化蛋白激酶MAPK1(ERK2)的磷酸化水平,从而干扰MAPK/ERK信号通路,该通路调控细胞生长和分化。
5. 调节激素相关通路
在激素依赖性肿瘤如乳腺癌中,远志山酮III显示出对雌激素受体ESR1信号通路的干扰作用,并能抑制芳香化酶CYP19A1的活性。CYP19A1是雌激素合成的关键酶,其抑制可降低体内雌激素水平,从而抑制雌激素依赖性肿瘤的生长。
综上所述,远志山酮III通过同时作用于凋亡调控、信号转导、侵袭转移、DNA代谢及激素调节等多个关键节点,形成多靶点协同的抗肿瘤网络,这为其克服单靶点药物易产生耐药性的缺点提供了潜在优势。
尽管远志山酮III在体外显示出优异的抗肿瘤活性,但其能否开发成为药物,很大程度上取决于其成药性,包括药代动力学(ADME:吸收、分布、代谢、排泄)性质和安全性。
药代动力学(预测与初步研究)
* 吸收与口服生物利用度:如前所述,其高极性(低LogP,高TPSA)可能限制其通过被动扩散跨过胃肠道上皮细胞膜,预示其口服吸收可能较差,生物利用度可能较低。制剂技术(如纳米晶、脂质体、磷脂复合物)或结构修饰(制备前药)可能是改善其吸收的有效策略。
* 分布:预测其血脑屏障透过性低,主要分布于血液和全身循环系统,难以进入中枢神经系统。这对其用于脑肿瘤治疗是不利因素,但可能降低中枢副作用。其与血浆蛋白的结合率尚不明确,需实验测定。
* 代谢:远志山酮III本身是CYP2E1的抑制剂,但它自身也可能被其他CYP450同工酶(如CYP3A4)代谢。其糖苷结构可能在肠道或肝脏被β-葡萄糖苷酶水解,生成苷元,苷元的理化性质和活性可能与原型化合物有显著差异,这是其代谢研究中需要关注的重点。
* 排泄:预计其原型或代谢产物主要经肾脏(通过尿液)或胆汁(通过粪便)排泄。具体途径和速率有待体内实验确认。
安全性评价
初步的计算机预测显示其无明显的hERG通道抑制风险和强烈的遗传毒性信号(Ames试验值1.5)。然而,这不能替代全面的临床前安全性评价。未来需要系统进行急性毒性、长期毒性、生殖毒性等研究,以评估其安全窗口。其对CYP450酶的抑制活性也提示需要进行详细的药物-药物相互作用研究。
成药性挑战与优化方向
主要挑战在于其较差的膜渗透性和可能较低的口服生物利用度。未来的研究应侧重于:
1. 深入的体内药代动力学研究:在动物模型(小鼠、大鼠)中全面评估其ADME特性。
2. 制剂学研究:开发新型给药系统,如纳米递送系统,以提高其溶解性、稳定性和靶向性。
3. 结构优化:在保留核心药效团的前提下,对其进行合理的化学修饰,以改善其脂溶性和药代动力学性质。
远志山酮III作为一种具有多靶点抗肿瘤活性的天然化合物,其临床应用前景广阔,但也面临诸多挑战。
潜在应用方向:
1. 抗肿瘤辅助治疗或联合用药:鉴于其多靶点特性和逆转耐药的潜力,远志山酮III有望与现有化疗药物(如拓扑异构酶抑制剂、紫杉醇类等)或靶向药物联合使用,起到协同增效、降低剂量、克服耐药的作用。
2. 针对特定靶点异常活跃的肿瘤:对于STAT3持续激活、MCL1/BCL2高表达、或HIF1A信号通路亢进的肿瘤亚型,远志山酮III可能具有更精准的治疗潜力。
3. 开发为化学预防剂:其CYP2E1抑制和抗氧化活性,使其在由特定毒物或代谢异常引发的肿瘤化学预防领域可能具有一定价值。
4. 神经系统疾病:虽然BBB透过性差,但通过制剂技术改造或针对外周神经炎症相关疾病,仍可能挖掘其在神经保护方面的应用。
未来研究展望:
1. 机制深度挖掘:利用化学生物学手段(如化学蛋白质组学)寻找其直接作用靶点,阐明其最初的作用分子事件。
2. 体内药效学验证:在多种人源肿瘤异种移植(PDX)或转基因小鼠模型上,确证其体内抗肿瘤效果及对转移的抑制作用。
3. 药代动力学与制剂攻关:这是推动其走向临床的核心环节。必须解决其生物利用度低的问题,并明确其体内代谢图谱。
4. 安全性系统评价:完成规范的临床前安全药理学和毒理学研究。
5. 结构衍生化与构效关系研究:合成一系列衍生物,明确其药效团,以期获得活性更强、成药性更优的候选化合物。
远志山酮III是从传统中药远志中分离得到的具有显著生物活性的呫吨酮类化合物。从最初发现的CYP450酶抑制活性,到如今备受关注的广谱抗肿瘤作用,其研究价值不断被揭示。现有研究表明,远志山酮III通过协同作用于MCL1、BCL2、STAT3、HIF1A、MMP2等多个肿瘤发生发展中的关键靶点,诱导凋亡、抑制增殖与侵袭,展现出多途径抗肿瘤的独特优势。然而,其较高的极性和可能存在的药代动力学缺陷,是制约其向临床应用转化的主要瓶颈。未来研究需在深入阐明其分子机制的基础上,着力解决其成药性问题,并通过合理的药物化学修饰或先进制剂技术,优化其药学性质。远志山酮III的研究不仅为开发新型多靶点抗肿瘤药物提供了有前景的先导化合物,也是中药现代化研究中的一个典型案例,体现了从传统药用植物中挖掘现代创新药物的巨大潜力。

版权所有:© 成都普瑞法科技开发有限公司(2015)备案号:蜀ICP备15035167号-1 客服热线:400-829-7929
技术支持:南京库价















