
Product name: 惕各酰戈米辛H
Synonym name: Tigloylgomisin H
Catalogue No.: SBP04216
Cas No.: 66069-55-4
Formula: C28H36O8
Mol Weight: 500.588
Botanical Source:
Physical Description:
Type of Compound: Lignans
Purity: 95%~99%
Analysis Method: HPLC-DAD or/and HPLC-ELSD
Identification Method: Mass, NMR
Packing: Brown vial or HDPE plastic bottle
The product could be supplied from milligrams to grams. Inquire for bulk scale.
We provide solution to improve the water-solubility of compounds, thereby facilitating the variety of activity tests and clinic uses.
For Reference Standard and R&D, Not for Human Use Directly.
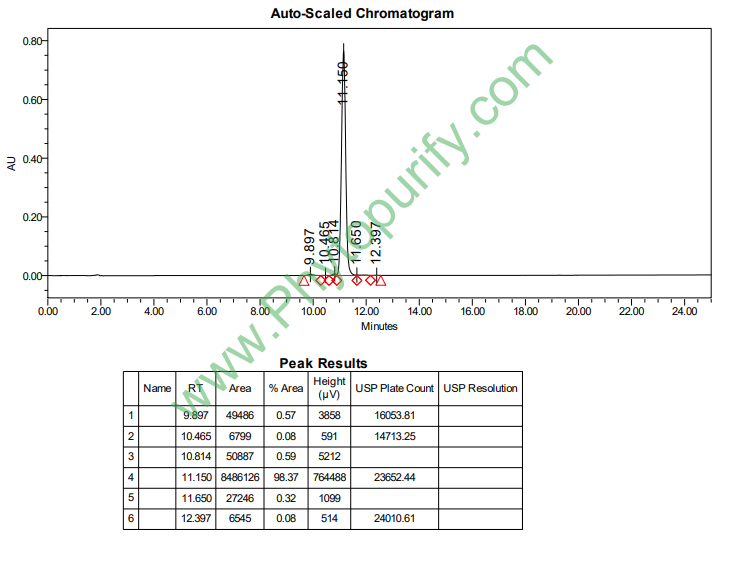
惕各酰戈米辛H的HPLC图谱
储存条件:短期保存 2~8℃,长期保存 -20 ~ -80℃
92.68
4.41
4.41
0.01
3.15
9.99
高
90.18
3.78
否
否
是
否
无
有
0.0
是
是
是
是

在天然产物化学与药理学研究领域,木酚素类化合物因其广泛的生物活性而备受关注。惕各酰戈米辛H(Tigloylgomisin H, CAS号:66069-55-4)是从传统中药五味子(Schisandra chinensis)果实中分离得到的一种联苯环辛烯类木酚素。五味子素有“安神、益气、生津”之功效,现代药理学研究揭示其活性成分在保肝、抗氧化、抗炎及抗肿瘤等方面具有显著潜力。惕各酰戈米辛H作为其中一员,其独特的生物活性谱,特别是作为单功能诱导剂选择性激活II相解毒酶的能力,使其在化学预防领域,尤其是肝癌的预防中,展现出重要的研究价值。本文旨在系统综述惕各酰戈米辛H的化学结构、植物来源、药理活性、作用机制、成药性评价及临床应用前景,以期为该化合物的深入研究和开发提供全面的科学参考。
惕各酰戈米辛H属于联苯环辛烯类木酚素,其分子式为C28H34O8,分子量为500.5880。其基本骨架由两个苯丙烷单元通过侧链β-碳原子连接构成一个联苯结构,并形成一个环辛二烯环。惕各酰戈米辛H的结构特征在于其C-6或C-7位(具体位置取决于立体化学)上连接有一个惕各酰氧基(tigloyloxy)取代基,该基团为其重要的活性基团之一。惕各酰酸(2-甲基-2-丁烯酸)的引入显著影响了化合物的亲脂性和与靶蛋白的相互作用能力。
从理化性质分析,该化合物具有较高的脂溶性,计算LogP值为4.4122,表明其易于穿透细胞膜,但水溶性较差(约0.0070 mg/mL),这为其制剂开发带来一定挑战。其拓扑极性表面积(TPSA)为92.68 Ų,属于中等极性分子。初步的成药性风险评估显示,惕各酰戈米辛H具有较高的血脑屏障透过潜力,这暗示其可能对中枢神经系统相关疾病具有潜在作用,但同时也需关注其在中枢的潜在效应。重要的是,该化合物在初步筛选中未显示hERG钾通道抑制活性(hERG抑制:否),且Ames试验结果为0.0,提示其致突变风险较低,具有相对良好的安全性起点。
惕各酰戈米辛H主要来源于五味子科五味子属植物五味子(Schisandra chinensis (Turcz.) Baill.)的干燥成熟果实。五味子主要分布于中国东北、韩国、日本及俄罗斯远东地区,其果实富含多种木酚素类成分,如五味子醇、五味子酯、戈米辛系列等,惕各酰戈米辛H是其中含量相对较低但活性显著的一个组分。
其提取分离通常遵循天然产物化学的常规流程。首先,将干燥的五味子果实粉碎,采用甲醇、乙醇或丙酮等有机溶剂进行回流提取或超声辅助提取。获得的粗提物经减压浓缩后,依次用石油醚、乙酸乙酯、正丁醇等溶剂进行梯度萃取,惕各酰戈米辛H主要富集于乙酸乙酯萃取部位。进一步的纯化多依赖于多种色谱技术组合,包括硅胶柱层析(以氯仿-甲醇或石油醚-乙酸乙酯系统梯度洗脱)、反相硅胶柱层析(如ODS, 以甲醇-水系统洗脱)、以及高效液相色谱(HPLC)制备。通过核磁共振(NMR, 包括1H-NMR、13C-NMR、2D-NMR)、质谱(MS)及与文献数据对比,可最终鉴定其化学结构。现代绿色提取技术如超临界CO2流体萃取,因其选择性高、无溶剂残留,也正被探索用于此类高价值木酚素的高效获取。
惕各酰戈米辛H的药理活性研究目前虽处于临床前阶段,但已揭示出其在癌症化学预防和抗炎方面的突出潜力。
癌症化学预防活性:该化合物最显著的特征是能够作为单功能诱导剂,特异性诱导II相解毒酶。研究表明,惕各酰戈米辛H能有效诱导小鼠肝癌细胞Hepa1c1c7中醌还原酶(QR, 即NAD(P)H:醌氧化还原酶1, NQO1)的活性。NQO1是细胞内关键的抗氧化和解毒酶,能将具有致癌潜力的醌类物质还原为氢醌,促进其随后的结合与排泄,从而保护细胞免受氧化损伤和致癌物攻击。惕各酰戈米辛H的这种诱导作用具有高度选择性,不诱导I相代谢酶(如细胞色素P450),避免了可能将前致癌物活化为终致癌物的风险,这使其成为一种理想的化学预防剂候选物,尤其适用于肝癌的预防。
抗炎活性:除了解毒酶诱导作用,惕各酰戈米辛H在多种炎症模型中也显示出明确的抗炎效应。其抗炎作用涉及对多个关键炎症介质和信号通路的调控。研究表明,该化合物能够抑制脂多糖(LPS)等刺激因子诱导的炎症因子如肿瘤坏死因子-α(TNF-α)、白细胞介素-6(IL-6)以及诱导型一氧化氮合酶(NOS2)的表达。此外,它还能抑制环氧合酶-1(PTGS1/COX-1)的活性,减少前列腺素类炎症介质的生成。在疼痛相关的神经源性炎症中,惕各酰戈米辛H对瞬时受体电位香草酸亚型1(TRPV1)和锚定蛋白亚型1(TRPA1)通道的潜在调节作用也值得关注。这些广泛的抗炎靶点提示其在治疗慢性炎症性疾病,如关节炎、结肠炎及神经炎症等方面具有应用前景。
惕各酰戈米辛H发挥其药理作用的核心机制在于对细胞防御系统的精准调控,其分子靶点网络主要围绕抗氧化应激反应元件(ARE)和炎症信号通路。
Nrf2-ARE通路的核心作用:惕各酰戈米辛H作为单功能诱导剂,其诱导NQO1等II相解毒酶的核心机制是通过激活核因子E2相关因子2(Nrf2)信号通路实现的。在静息状态下,Nrf2与其抑制蛋白Keap1结合于细胞质中,并被泛素化降解。惕各酰戈米辛H可能通过修饰Keap1上的关键半胱氨酸残基,或影响Nrf2与Keap1的相互作用,促使Nrf2从Keap1上解离并稳定。随后,Nrf2易位至细胞核,与抗氧化反应元件(ARE)结合,启动下游一系列细胞保护基因的转录,包括NQO1、谷胱甘肽S-转移酶(GST)、血红素加氧酶-1(HO-1)等。这一通路是细胞抵抗氧化应激和亲电性毒物的核心防御机制,也是惕各酰戈米辛H发挥化学预防作用的分子基础。
抗炎作用的多元靶点:其抗炎作用涉及对多条经典炎症信号通路的干预:
这些机制并非孤立存在,Nrf2通路的激活本身也具有强大的抗炎效应,因为HO-1等产物能抑制促炎介质的产生。因此,惕各酰戈米辛H通过协同激活保护性通路(Nrf2)和抑制破坏性通路(NF-κB、STAT3等),构建了一个多靶点、网络化的药理作用模式。
基于其理化参数和初步生物学数据,对惕各酰戈米辛H的成药性进行初步评价。
类药性与吸收分布:分子量(500.6)略高于“类药五规则”的常规上限(500),但仍在可接受范围。较高的脂溶性(LogP > 4)预示其口服生物利用度可能面临挑战,因为过高的亲脂性可能导致在胃肠道内容物中溶解性差,或在肠壁和肝脏中经历强烈的首过代谢。其高血脑屏障透过性(预测)提示其能进入中枢,这对中枢相关疾病的治疗可能是优势,但也需评估潜在的中枢副作用。目前缺乏其体内吸收、分布、代谢、排泄(ADME)的详细实验数据。
代谢与安全性:作为外源性物质,惕各酰戈米辛H在体内很可能主要经肝脏细胞色素P450酶系代谢,其惕各酰基团和木酚素骨架上的甲氧基、羟基等都可能成为代谢位点,生成葡萄糖醛酸或硫酸结合物。其不抑制hERG通道是一个重要的安全性利好,降低了引发心脏QT间期延长和尖端扭转型室性心动过速的风险。Ames试验阴性初步排除了其直接的基因毒性,但完整的遗传毒性组合试验和长期毒理学研究仍有待开展。
药代动力学研究缺口:目前公开的文献中,关于惕各酰戈米辛H系统的药代动力学研究,包括其绝对生物利用度、血浆蛋白结合率、组织分布特征、主要代谢产物鉴定、消除半衰期等关键参数,几乎处于空白状态。这是推进其向药物开发迈进必须填补的核心信息缺口。利用液相色谱-串联质谱(LC-MS/MS)技术建立高灵敏度的体内分析方法,是开展这些研究的前提。
惕各酰戈米辛H展现出的独特药理特性为其临床应用指明了潜在方向,但同时也面临着从先导化合物到候选药物的典型挑战。
潜在应用领域:
面临的挑战与未来研究方向:
惕各酰戈米辛H作为源自传统中药五味子的联苯环辛烯类木酚素,凭借其通过Nrf2-ARE通路特异性诱导II相解毒酶NQO1的独特能力,以及在NF-κB、STAT3等多条炎症通路上的抑制作用,确立了其在癌症化学预防和抗炎治疗领域的显著研究价值。其作为“单功能诱导剂”的特性,在增强细胞防御的同时避免了潜在的风险,体现了天然产物在调控复杂生物网络中的智慧。尽管目前其在成药性,尤其是溶解性和系统药代动力学方面存在明显短板,但这些挑战也正是现代药物化学和药剂学可以着力解决的环节。未来,通过多学科交叉合作,深入揭示其分子作用细节,优化其理化性质,并完成系统的临床前开发,惕各酰戈米辛H有望从一个有潜力的天然先导化合物,发展成为用于预防和治疗肝脏疾病及炎症相关疾病的创新药物或健康产品,为人类健康贡献来自传统医学的现代力量。

版权所有:© 成都普瑞法科技开发有限公司(2015)备案号:蜀ICP备15035167号-1 客服热线:400-829-7929
技术支持:南京库价















