
产品名称: 去甲波尔定
英文名: Norboldine
中文别名: 去甲波尔定;新木姜子碱
英文别名: Laurolistine;Laurolitsine;Norisoboidine;(+)-Norboldine
Cas 号: 5890-18-6(游离)
产品编码:BP2011
分子式: C18H19NO4
分子量: 349.11
来源:
物理性状:
化合物类型:
纯度: 95%~99%
分析方法: HPLC-DAD or/and HPLC-ELSD
鉴定方法: 质谱(Mass), 核磁(NMR)
包装: 棕色小玻璃瓶,标准包装10mg,20mg,50mg;可以按客户需求包装。
可以满足克级以上大量需求,详情请咨询。
提供相关图谱和分析方法指导,可以提供包括色谱柱,标准品和分析方法在内的含量测定整体服务包,价格优惠。
我们提供改善产品水溶性解决方案,改善化合物在水中的溶解度,便于进行各种活性试验和临床应用。
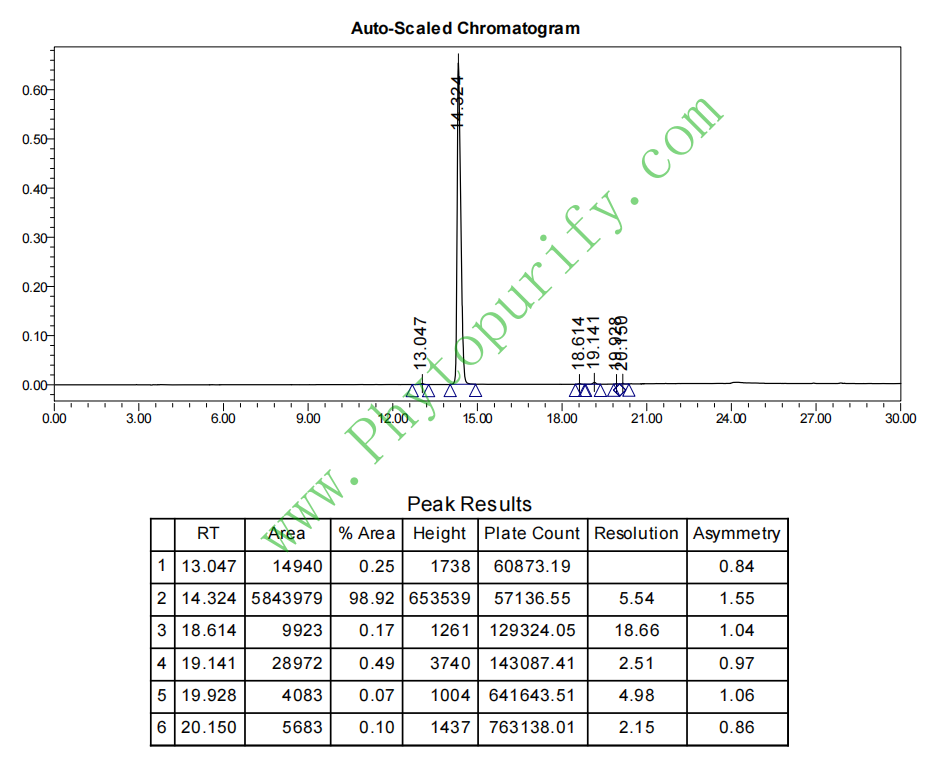
去甲波尔定的HPLC图谱
储存条件:短期保存 2~8℃,长期保存 -20 ~ -80℃
71.04
1.70
1.70
低
否

去甲波尔定(Norboldine,CAS号:5890-18-6)是一种具有独特结构特征的阿朴啡类生物碱,最初从木姜子(Lindera aggregata)和山药(Dioscorea spp.)等植物中分离得到。作为去甲吗啡的衍生物,去甲波尔定在其分子结构上具备2位和9位羟基以及1位和10位甲氧基的取代特征,赋予其独特的生物活性。近年来,随着天然产物药理学的深入发展,去甲波尔定因其对HIV-1整合酶的抑制作用而备受关注,显示出潜在的抗病毒药物开发价值。此外,其作为阿朴啡类生物碱的代表,在抗炎、抗肿瘤及神经保护等多方面的药理活性研究中展现出广阔的应用前景。
本文旨在系统综述去甲波尔定的化学结构与理化性质、植物来源及提取方法,深入探讨其药理活性及作用机制,结合成药性评价与药代动力学特征,展望其在临床应用中的潜力与未来发展方向,为该天然产物的药物开发提供理论依据与研究参考。
去甲波尔定属于阿朴啡(aporphine)类生物碱,分子式C19H23NO4,分子量为313.35。其结构特征为去甲吗啡骨架的2位和9位羟基(-OH)取代,以及1位和10位甲氧基(-OCH3)取代,形成酚类和芳香族醚的功能团。该结构赋予其较高的极性,TPSA(拓扑极表面积)为71.04 Ų,表明其具有一定的氢键受体能力(5个氢键受体),有利于与生物靶点形成稳定的相互作用。
去甲波尔定的LogP值为1.7,显示出适中的脂溶性,利于细胞膜的穿透但血脑屏障渗透性较低(Blood-Brain Barrier渗透性为Low),提示其在中枢神经系统的药效发挥可能受限。其理化性质稳定,适合于药物制剂的开发,但肝毒性、心脏毒性、hERG通道抑制及致突变性(Ames试验)等安全性指标尚未明确,需要进一步系统评价。
去甲波尔定主要分布于木姜子属(Lindera spp.)和部分山药属(Dioscorea spp.)植物中。木姜子作为传统中药材,广泛用于治疗消化系统疾病及炎症,其根茎和叶片中含有丰富的阿朴啡类生物碱。山药则以其药食两用的特性著称,部分种类中也检测到去甲波尔定的存在。
提取去甲波尔定的常用方法包括:
溶剂提取:采用甲醇、乙醇或甲醇-水混合溶剂对植物干燥粉末进行回流提取,利用其极性特征有效溶解目标生物碱。
液液分配:通过调节pH值,将生物碱从水相转移至有机相(如氯仿或乙酸乙酯),实现初步纯化。
柱层析分离:利用硅胶或C18反相柱进行分离纯化,结合梯度洗脱技术,获得高纯度的去甲波尔定。
高效液相色谱(HPLC)分析:用于定性定量检测,确保提取物的质量和纯度。
近年来,超声辅助提取和微波辅助提取技术也被引入,提高了提取效率和纯度,降低了溶剂用量和提取时间,为工业化生产提供了技术支持。
去甲波尔定最引人注目的药理活性是其对HIV-1整合酶(IN)的抑制作用。HIV-1整合酶是病毒复制周期中的关键酶,负责将病毒DNA整合入宿主基因组,是抗逆转录病毒治疗的重要靶点。体外实验表明,去甲波尔定能够有效抑制HIV-1整合酶活性,阻断病毒基因组的整合过程,从而抑制病毒复制。
此外,去甲波尔定对逆转录酶(RT)、蛋白酶(PR)等其他HIV关键酶的影响尚需进一步研究,但其作为天然产物的多靶点潜力为抗HIV药物开发提供了新的思路。
阿朴啡类生物碱普遍具有抗炎作用,去甲波尔定通过抑制核因子κB(NF-κB)信号通路及环氧合酶-2(COX-2,PTGS2)表达,减少炎症介质如肿瘤坏死因子α(TNF-α)的释放,表现出显著的抗炎效果。这一作用机制为其在炎症性疾病如关节炎、炎症性肠病中的应用提供了理论基础。
初步研究显示,去甲波尔定对多种肿瘤细胞系具有抑制增殖和诱导凋亡的作用。其机制可能涉及调节p53蛋白(TP53)功能,抑制抗凋亡蛋白Bcl-2(BCL2)的表达,以及阻断表皮生长因子受体(EGFR)和血管内皮生长因子受体(KDR)信号通路,从而抑制肿瘤细胞的生长和血管生成。
尽管去甲波尔定血脑屏障渗透性较低,但其对乙酰胆碱酯酶(ACHE)的抑制作用及对β-淀粉样蛋白前体蛋白(APP)和α-突触核蛋白(SNCA)相关病理过程的调节,提示其在神经退行性疾病如阿尔茨海默病和帕金森病中的潜在应用价值。
去甲波尔定的药理作用依赖于其与多种分子靶点的相互作用,具体机制如下:
HIV-1整合酶抑制:去甲波尔定通过与整合酶的活性位点结合,阻断病毒DNA与宿主染色质的整合过程,抑制病毒复制。分子对接和动力学模拟显示其羟基和甲氧基基团参与关键氢键和疏水相互作用,增强结合亲和力。
抗炎信号通路调控:通过抑制NF-κB的核转位,减少促炎细胞因子(如TNF-α、IL-6)的表达;同时抑制COX-2酶活性,降低前列腺素合成,减轻炎症反应。
肿瘤相关信号通路干预:调节p53介导的细胞周期和凋亡通路,降低Bcl-2表达,促进肿瘤细胞凋亡;同时抑制EGFR和KDR信号,阻断肿瘤增殖和血管生成。
神经保护机制:抑制乙酰胆碱酯酶活性,延长乙酰胆碱作用时间,改善认知功能;调节APP和SNCA蛋白代谢,减缓神经毒性蛋白聚集。
去甲波尔定的成药性参数显示其具有一定的药物开发潜力:
然而,去甲波尔定的血脑屏障渗透性较低,限制了其在中枢神经系统疾病中的直接应用。安全性方面,肝毒性、心脏毒性及hERG通道抑制等关键指标尚未明确,需通过系统的体内外毒理学研究进行评估。
药代动力学数据目前较为缺乏,未来研究应重点关注其吸收、分布、代谢及排泄(ADME)特性,尤其是代谢途径及代谢产物的活性与安全性评估。
去甲波尔定作为一种具有多靶点作用的天然阿朴啡类生物碱,展现出广泛的药理活性,尤其是在抗HIV领域具有显著潜力。其对HIV-1整合酶的抑制作用为开发新型抗逆转录病毒药物提供了重要的化学骨架和先导化合物。此外,去甲波尔定的抗炎、抗肿瘤及神经保护作用拓宽了其临床应用的可能性。
未来研究应重点开展:
结构优化与衍生物设计:通过化学修饰提高其靶向活性和药代动力学性能,解决血脑屏障渗透性差及潜在毒性问题。
系统的药理毒理评价:明确其安全性,尤其是肝脏和心脏毒性,确保临床应用的安全性。
体内药效与机制验证:利用动物模型验证其抗病毒、抗炎及抗肿瘤效果,进一步阐明分子机制。
多靶点联合治疗策略:结合其他抗病毒或抗肿瘤药物,发挥协同效应,提高治疗效果。
临床前及临床研究:推动去甲波尔定及其衍生物进入临床试验阶段,评估其药效和安全性。
去甲波尔定作为一种天然来源的阿朴啡类生物碱,凭借其独特的化学结构和多样的药理活性,尤其是对HIV-1整合酶的抑制作用,展现出重要的药物开发价值。尽管目前其成药性和安全性数据尚不完善,但随着现代药物化学和药理学技术的发展,去甲波尔定有望成为抗病毒及多种疾病治疗的新型候选药物。未来通过系统的结构优化、机制研究及临床评价,去甲波尔定有望在天然产物药物开发领域占据一席之地,推动相关疾病治疗策略的创新与进步。

版权所有:© 成都普瑞法科技开发有限公司(2015)备案号:蜀ICP备15035167号-1 客服热线:400-829-7929
技术支持:南京库价















