
产品名称: 夹竹桃苷A
英文名: Oleaside A
中文别名:
英文别名:
Cas 号: 69686-84-6
产品编码:BP3283
分子式: C30H44O7
分子量: 516.675
来源:
物理性状:
化合物类型: Triterpenoids
纯度: 95%~99%
分析方法: HPLC-DAD or/and HPLC-ELSD
鉴定方法: 质谱(Mass), 核磁(NMR)
包装: 棕色小玻璃瓶,标准包装10mg,20mg,50mg;可以按客户需求包装。
可以满足克级以上大量需求,详情请咨询。
提供相关图谱和分析方法指导,可以提供包括色谱柱,标准品和分析方法在内的含量测定整体服务包,价格优惠。
提供改善产品水溶性解决方案,改善化合物在水中的溶解度,便于进行各种活性试验和临床应用。
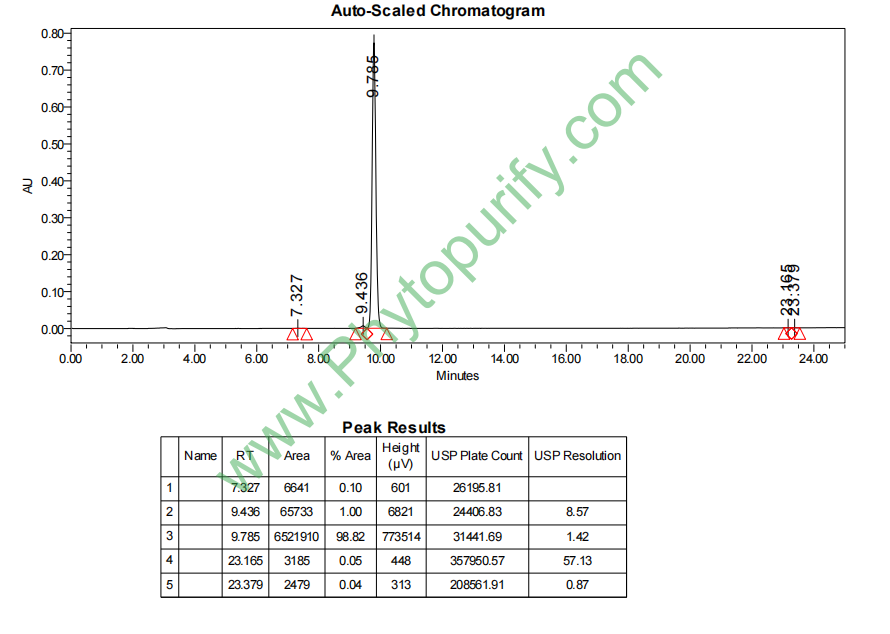
夹竹桃苷A的HPLC图谱
储存条件:短期保存 2~8℃,长期保存 -20 ~ -80℃
91.29
3.25
3.25
0.01
2.39
10.02
高
75.04
6.31
否
否
否
否
有
无
0.3
否
否
否
否

夹竹桃苷A(Oleaside A),化学名称为一种极性烯醇内酯单糖苷,是一种从夹竹桃(Nerium oleander)中分离得到的天然活性化合物。其CAS号为69686-84-6,分子式为C30H44O7,分子量约为516.68 g/mol。作为一种结构独特的天然产物,夹竹桃苷A自被发现以来,便因其显著的生物活性而受到天然产物药学与药物化学领域的关注。现有研究初步表明,该化合物能够抑制白细胞介素-1α(IL-1α)和肿瘤坏死因子-α(TNF-α)对细胞间粘附分子-1(ICAM-1)的诱导作用,显示出潜在的抗炎与抗肿瘤活性。其作用靶点主要指向钠钾ATP酶(Na+/K+-ATPase)的多个亚型(ATP1A1, ATP1A2, ATP1B1, ATP1B2, ATP1B3),这一特性将其药理作用与心力衰竭等疾病关联起来。本文将从其化学结构、植物来源、药理机制、成药性评估及研究前景等方面,对这一具有研究价值的天然产物进行系统性的专业科普介绍。
夹竹桃苷A的分子式为C30H44O7,属于中等分子量的天然产物。其SMILES结构式为:COC1CC(OC2CCC3(C)C(CCC45CCC(C6=CC(=O)OC6)C(C)(CCC43)C5=O)C2)OC(C)C1O。从结构式解析,该分子包含一个烯醇内酯环、一个糖苷结构(单糖部分)以及多个甲基、甲氧基和羟基取代基,属于极性较大的苷类化合物。这种结构特征决定了其独特的理化性质。
根据提供的成药性参数,其脂水分配系数对数(LogP)为3.2494,表明该化合物具有一定的亲脂性,但并非高度疏水。拓扑极性表面积(TPSA)为91.29 Ų,这个数值相对适中,通常与化合物透过生物膜的能力相关。分子量(MW)516.68略高于常见口服药物500 Da的经验阈值,这是其成药性评估中需要关注的一点。水溶性参数为0.0119(单位未明确,通常表示溶解度较低),这与LogP值显示的亲脂性趋势一致。Caco-2细胞渗透性为10.0156,提示其可能具有较好的肠道吸收潜力。值得注意的是,其血脑屏障(BBB)穿透性被预测为“高”,这意味着该化合物有可能进入中枢神经系统,这对于作用于中枢靶点的药物开发是优势,但也可能带来潜在的神经毒性风险。血浆蛋白结合率(PPB)为75.04%,属于中等偏高水平,可能影响其游离药物浓度和药效。
夹竹桃苷A的植物来源单一且明确,即夹竹桃(Nerium oleander L.)。夹竹桃属于夹竹桃科(Apocynaceae),是一种常绿灌木,原产于地中海地区,现广泛分布于全球热带和亚热带地区作为观赏植物。值得注意的是,夹竹桃全株有毒,其毒性主要源于所含的多种强心苷类化合物(如欧夹竹桃苷),这些成分能强烈抑制心肌细胞上的Na+/K+-ATP酶,导致心律失常甚至死亡。
在传统医学中,夹竹桃的应用历史悠久但充满风险。在印度阿育吠陀医学和某些民间疗法中,夹竹桃的叶、皮、根等部位曾被极小剂量地用于治疗心脏病、哮喘、癫痫、麻风病等。然而,由于其治疗窗极窄(有效剂量与中毒剂量非常接近),使用不当极易导致严重中毒,因此现代正规医学已严格限制其直接药用。夹竹桃苷A的发现,代表了现代天然产物化学从传统有毒植物中“去粗取精”,寻找特定、有效且可能毒性较低的单一活性成分的研究思路。它不同于经典的强心苷,是一种极性烯醇内酯单糖苷,其毒性特征和药理靶点可能与已知强心苷有所不同,这为安全开发利用夹竹桃资源提供了新的科学切入点。
夹竹桃苷A的药理活性研究目前仍处于早期阶段,但已有的数据揭示了其多方面的潜力。
4.1 抗炎与免疫调节活性
现有中文描述明确指出,夹竹桃苷A“可抑制IL-1α和TNF-α对ICAM-1的诱导作用”。IL-1α和TNF-α是两种关键的促炎细胞因子,在炎症反应和免疫应答中扮演核心角色。它们能诱导血管内皮细胞、上皮细胞等多种细胞表面表达ICAM-1。ICAM-1是一种重要的细胞粘附分子,介导白细胞与血管内皮细胞的粘附和迁移,是炎症细胞浸润组织的关键步骤。因此,抑制ICAM-1的诱导表达,意味着夹竹桃苷A可能通过干预这一早期炎症信号通路,发挥抗炎作用。这种机制在治疗由慢性炎症驱动的疾病,如动脉粥样硬化、类风湿性关节炎以及某些肿瘤(肿瘤微环境中存在慢性炎症)中具有潜在应用价值。
4.2 潜在的抗肿瘤活性
描述中提及的“具有抗肿瘤活性”可能与其抗炎机制间接相关,因为慢性炎症是肿瘤发生和发展的促进因素。此外,更直接的作用可能与其靶向Na+/K+-ATP酶有关。研究表明,某些Na+/K+-ATP酶抑制剂不仅能影响离子稳态,还能通过激活下游信号通路(如Src激酶、MAPK通路)来影响细胞增殖、凋亡和迁移,从而在特定肿瘤模型中显示出抗肿瘤效应。夹竹桃苷A对多个ATP酶亚型的抑制作用,可能为其抗肿瘤研究提供了理论基础,但具体的作用通路和细胞模型数据仍需进一步实验证实。
4.3 核心作用靶点:Na+/K+-ATP酶
数据库提供的5个明确靶点(ATP1A1, ATP1A2, ATP1B1, ATP1B2, ATP1B3)全部是Na+/K+-ATP酶(又称钠泵)的亚基。Na+/K+-ATP酶是一种存在于几乎所有动物细胞膜上的跨膜蛋白,其功能是通过水解ATP,将细胞内的3个Na+泵出,同时将2个K+泵入,从而维持细胞膜两侧的钠钾离子梯度。这一梯度对于维持细胞渗透压、神经冲动传导、心肌细胞静息电位和收缩力至关重要。
- ATP1A1、ATP1A2:是催化α亚基的两种异构体。α1亚基(ATP1A1)广泛表达,α2亚基(ATP1A2)主要在心脏、骨骼肌和大脑中表达。
- ATP1B1、ATP1B2、ATP1B3:是调节β亚基的三种异构体,主要参与α亚基的正确折叠、膜定位和功能调节。
4.4 与心力衰竭(Heart Failure)的关联
夹竹桃苷A的靶点信息将其与“心衰”这一疾病直接关联。这正是基于其Na+/K+-ATP酶抑制剂的属性。临床上经典的强心药(如地高辛)正是通过抑制心肌细胞上的Na+/K+-ATP酶(主要靶向α亚基)来发挥正性肌力作用。其机制是:抑制钠泵后,细胞内Na+浓度升高,进而通过Na+/Ca2+交换体(NCX)减少Ca2+外排或增加Ca2+内流,导致细胞内Ca2+浓度增加,从而增强心肌收缩力。因此,从作用靶点推测,夹竹桃苷A可能具有类似的强心潜力。然而,其具体对哪个亚型的选择性更高、强心效价与毒性(特别是致心律失常风险)的比例如何,是决定其能否成为新型抗心衰候选药物的关键。数据库将其列为相关疾病,提示了该研究方向,但需要严格的临床前心血管药理学评估。
基于提供的成药性参数,我们可以对夹竹桃苷A作为潜在药物的开发前景进行初步评估,并参考著名的“Lipinski五规则”(Rule of Five,RO5)进行对照分析。RO5通常用于预测小分子化合物的口服吸收特性,其内容为:分子量MW < 500 Da,氢键供体数HBD < 5,氢键受体数HBA < 10,脂水分配系数LogP < 5。
综合评估:夹竹桃苷A在渗透性、脂溶性、部分毒性指标上表现尚可,但其分子量略高和存在染色体畸变风险是两个主要的成药性障碍。特别是染色体畸变阳性,是临床前开发中需要重点攻关和明确机理的“红旗”信号。它必须通过更深入的遗传毒性测试(如微核试验、彗星试验等)来确认,并评估其是否具有阈值效应,以及是否可通过结构修饰消除。其Na+/K+-ATP酶抑制活性也要求进行详尽的心血管安全药理评价。
目前,关于夹竹桃苷A的公开研究文献相对有限,其活性数据多来源于初步的筛选和数据库收录信息。它代表了一类从有毒植物夹竹桃中发现的、结构不同于传统强心苷的新型活性分子,其研究价值在于探索夹竹桃化学成分多样性与药理活性的新维度。
研究现状:
1. 基础研究阶段:目前的研究主要集中在化合物的分离鉴定、初步的靶点预测和细胞水平活性筛选。其抑制ICAM-1诱导的抗炎机制和Na+/K+-ATP酶靶向性为其主要药理标签,但深入的信号通路研究、动物模型验证数据仍然缺乏。
2. 结构-活性关系(SAR)空白:作为单一化合物,其化学结构与其他已知药物的相似性较低,尚未见系统的衍生物合成与活性优化研究。
3. 安全性评估亟待深入:如前所述,染色体畸变的潜在风险是悬在其开发之路上的“达摩克利斯之剑”,必须通过标准化的GLP毒理学研究来明确。
应用前景与未来方向:
1. 作为先导化合物:夹竹桃苷A的核心价值可能在于其独特的烯醇内酯单糖苷骨架。药物化学家可以以其为先导结构,进行系统的结构修饰与优化。目标包括:在保留或增强活性的前提下,降低分子量以改善类药性;通过化学修饰尝试消除染色体畸变活性;提高对特定Na+/K+-ATP酶亚型(如主要表达于心肌的α2/α1亚型)的选择性,以期获得强心作用强而神经系统副作用及心律失常风险低的新衍生物。
2. 机制驱动的疾病模型研究:未来研究应在其已预测的靶点和活性基础上,设计严谨的实验。例如:
- 抗炎/抗肿瘤方向:在明确的炎症或肿瘤细胞模型中,验证其对ICAM-1表达及白细胞粘附的影响,并探索其下游通路。
- 心血管方向:在离体心脏、心衰动物模型中,评估其正性肌力作用、治疗心衰的疗效以及治疗窗口,并与经典强心苷进行对比。
3. 深入毒理学与药代动力学研究:在开展任何疗效研究的同时,必须平行进行全面的临床前安全评价(尤其是遗传毒性和心血管毒性)以及药代动力学(吸收、分布、代谢、排泄)研究,以获取完整的开发可行性数据。
总之,夹竹桃苷A是一个具有明确生物活性靶点和独特化学结构的天然产物,为开发新型抗炎、抗肿瘤或心血管药物提供了有趣的起点。然而,其迈向候选药物之路充满挑战,特别是安全性方面的疑虑。未来的研究需要在严谨的科学设计下,平衡其活性潜力与成药性风险,通过多学科交叉合作,方能揭示其真正的转化医学价值。

版权所有:© 成都普瑞法科技开发有限公司(2015)备案号:蜀ICP备15035167号-1 客服热线:400-829-7929
技术支持:南京库价















