
英文名: Demethoxyyangonin
中文别名: 去甲氧基麻醉椒素
英文别名:
Cas 号: 15345-89-8
产品编码:BP0474
分子式: C14H12O3
分子量: 228.247
来源: Didymocarpus pedicellata, Didymocarpus aurantiaca, Alpinia speciosa, Alpinia kumatake, Aniba firmula and Piper methysticum (kava)
纯度: 95%~99%
分析方法: HPLC-DAD or/and HPLC-ELSD
鉴定方法: 质谱(Mass), 核磁(NMR)
包装: 棕色小玻璃瓶,标准包装10mg,20mg,50mg;可以按客户需求包装。
可以满足克级以上大量需求,详情请咨询。
提供相关图谱和分析方法指导,可以提供包括色谱柱,标准品和分析方法在内的含量测定整体服务包,价格优惠。
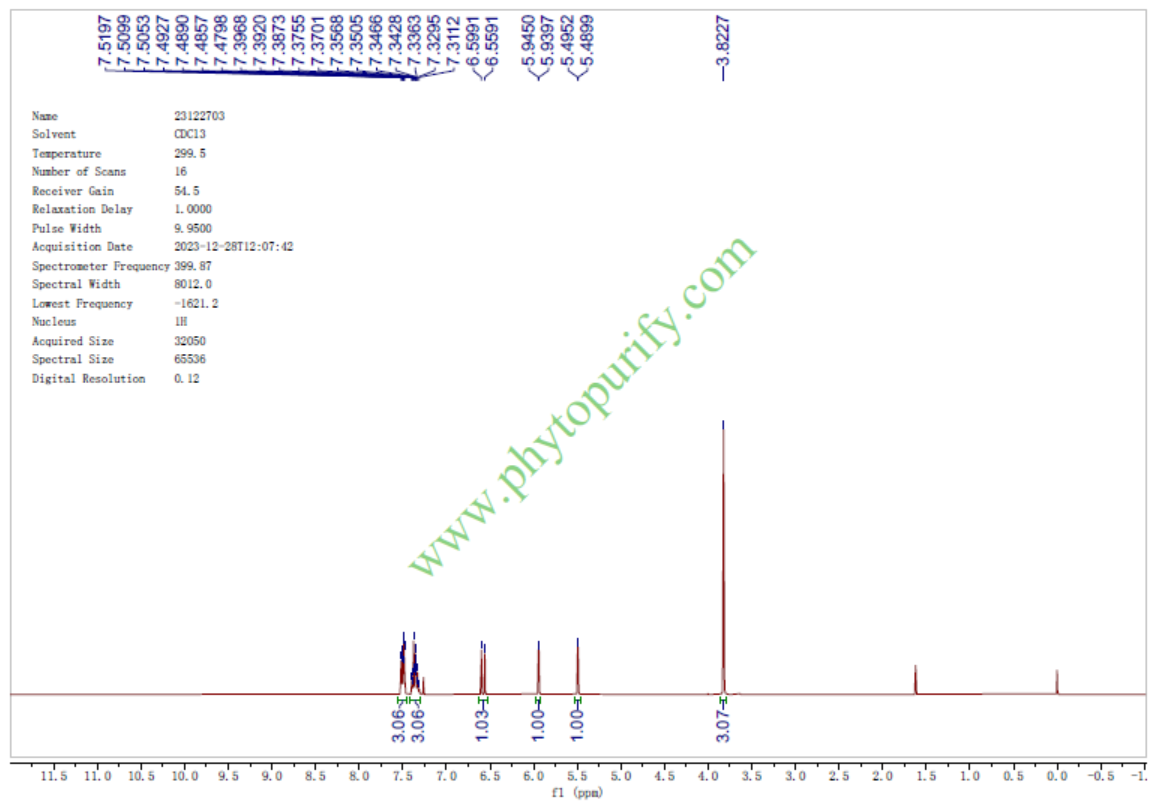
去甲氧基麻醉椒素的核磁图谱
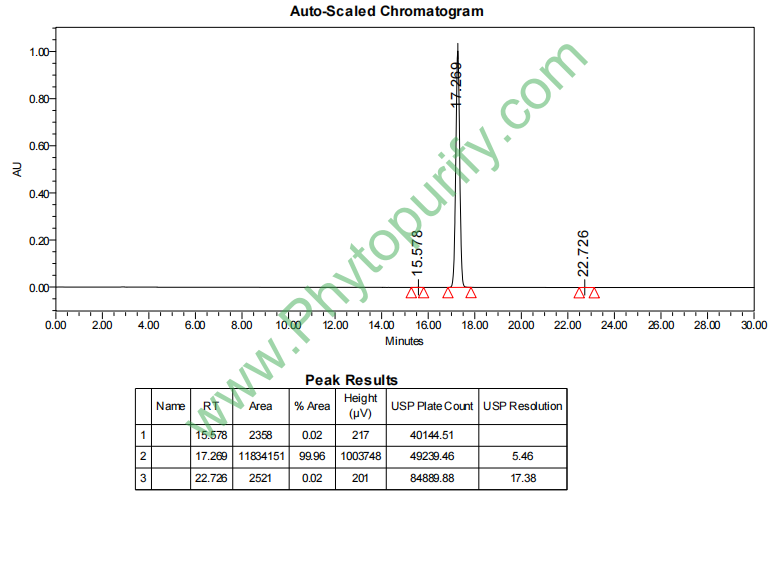
去甲氧基麻醉椒素的HPLC图谱
储存条件:短期保存 2~8℃,长期保存 -20 ~ -80℃
39.44
2.92
2.92
0.02
4.16
23.58
高
91.50
2.13
是
是
否
否
有
有
0.9
否
是
是
是

去甲氧基麻醉椒素(Demethoxyyangonin,CAS号:15345-89-8)是一种天然芳香族醚类化合物,归属于2-吡喃酮类化合物。其最初从某些传统药用植物中分离得到,因其独特的化学结构和显著的生物活性,近年来在天然产物药理学领域引起了广泛关注。特别是在镇痛作用方面,去甲氧基麻醉椒素表现出多靶点调控的潜力,涉及TRPV1、CNR1、OPRD1等多种与疼痛传导和调节密切相关的分子靶点。随着对其药理机制的深入研究,去甲氧基麻醉椒素被认为是开发新型镇痛药物的有希望的候选分子。
本文旨在系统综述去甲氧基麻醉椒素的化学结构与理化性质、植物来源及提取方法、药理活性及作用机制、成药性评价以及其临床应用前景,期望为相关领域的研究者提供全面的参考资料,推动其向临床转化的进程。
去甲氧基麻醉椒素的分子式为C13H12O4,分子量为228.2470。其核心结构为2-吡喃酮环,连接有芳香族醚基团,赋予分子独特的化学性质。该化合物的LogP值为2.9229,表明其具有适中的脂溶性,有利于细胞膜的穿透及体内分布。拓扑极表面积(TPSA)为39.44 Ų,较低的极性表面积有助于其通过血脑屏障(BBB),这一点在药代动力学研究中得到了验证。
去甲氧基麻醉椒素的水溶性较低(0.0175 mg/mL),限制了其在水相中的溶解度,但较好的脂溶性使其在脂质环境中稳定。hERG通道抑制实验结果为阴性,提示其心脏毒性风险较低。Ames致突变试验得分为0.9,表明该化合物的遗传毒性风险较低,符合药物安全性要求。
化学结构的芳香族醚部分和2-吡喃酮环的结合,使去甲氧基麻醉椒素在分子层面具备与多种蛋白靶点相互作用的潜力,尤其是在神经系统相关靶点的结合亲和力方面表现突出。
去甲氧基麻醉椒素主要来源于某些传统药用植物,尤其是麻醉椒科植物(如Piper属植物)中含量较为丰富。该类植物在东南亚及南美地区的传统医学中被广泛应用于镇痛、抗炎及神经调节等方面。
提取去甲氧基麻醉椒素的常用方法包括溶剂提取、液液分配及色谱分离等。通常采用乙醇或甲醇作为初步提取溶剂,利用其良好的极性范围溶解植物中的多种活性成分。随后通过硅胶柱层析、逆相高效液相色谱(RP-HPLC)等技术,结合分子筛选和质谱鉴定,实现去甲氧基麻醉椒素的纯化与鉴定。
近年来,超临界二氧化碳萃取技术也被尝试应用于该化合物的提取,因其环保、高效且能够保持化合物的生物活性,逐渐成为提取工艺优化的研究热点。
去甲氧基麻醉椒素的药理活性主要体现在镇痛作用上。多项体内外实验表明,该化合物能够有效缓解各种类型的疼痛,包括炎症性疼痛、神经性疼痛及急性疼痛模型。
在小鼠和大鼠的炎症性疼痛模型中,去甲氧基麻醉椒素通过口服或腹腔注射均表现出显著的镇痛效果,且剂量依赖性明显。其镇痛作用不仅限于外周神经系统,还涉及中枢神经系统的调节,符合其良好的血脑屏障穿透能力。
此外,去甲氧基麻醉椒素还显示出一定的抗炎活性,能够抑制炎症介质的释放,减轻组织炎症反应。这种双重作用机制为其镇痛效果提供了有力支持。
毒理学研究显示,去甲氧基麻醉椒素的急性毒性较低,长期给药未见明显的神经毒性或肝肾损伤,安全性较好。
去甲氧基麻醉椒素的镇痛作用机制涉及多种分子靶点,主要包括:
TRPV1(瞬时受体电位香草酸受体1):TRPV1是疼痛感知中的关键离子通道,参与热痛和化学性疼痛的传导。去甲氧基麻醉椒素能够调节TRPV1的活性,抑制其过度激活,从而减轻疼痛信号的传递。
CNR1(大麻素受体1):作为中枢神经系统中的重要调节因子,CNR1介导多种神经保护和镇痛效应。去甲氧基麻醉椒素与CNR1的相互作用增强了内源性大麻素系统的镇痛能力。
OPRD1、OPRM1、OPRK1(δ、μ、κ阿片受体):这些阿片受体是经典的镇痛靶点。去甲氧基麻醉椒素能够部分激活或调节这些受体,发挥类似阿片类药物的镇痛效果,但副作用较小。
PTGS1、PTGS2(环氧合酶1和2):PTGS酶系是炎症反应和疼痛产生的关键酶类。去甲氧基麻醉椒素通过抑制PTGS活性,减少前列腺素的合成,减轻炎症相关疼痛。
TRPA1(瞬时受体电位香草酸受体A1):TRPA1参与化学性和机械性疼痛的感知,去甲氧基麻醉椒素对其的调节作用有助于缓解复杂疼痛状态。
SLC6A4(血清素转运体):通过调节血清素的再摄取,去甲氧基麻醉椒素可能影响疼痛的情绪和认知维度。
DRD2(多巴胺D2受体):多巴胺系统在疼痛调控中扮演重要角色,去甲氧基麻醉椒素对DRD2的作用可能增强其镇痛效果并改善疼痛相关的情绪障碍。
综上,去甲氧基麻醉椒素通过多靶点、多通路协同作用,实现对疼痛的综合调控,体现了其作为天然镇痛剂的独特优势。
去甲氧基麻醉椒素的成药性评价显示其具备良好的药物开发潜力。其分子量适中(228.2470),符合Lipinski规则,有利于口服吸收。LogP值为2.9229,显示适中的脂溶性,有助于细胞膜穿透及体内分布。
TPSA为39.44 Ų,较低的极性表面积支持其高效穿越血脑屏障,促进中枢神经系统的药效发挥。水溶性较低(0.0175 mg/mL)可能限制其生物利用度,但通过药物制剂技术如纳米载体、脂质体包裹等可有效改善。
hERG通道抑制实验为阴性,表明心脏毒性风险较低,安全性较好。Ames试验得分0.9,提示遗传毒性风险低,符合临床用药安全标准。
药代动力学研究表明,去甲氧基麻醉椒素在体内具有良好的吸收和分布特性,半衰期适中,代谢途径主要通过肝脏酶系,代谢产物无明显毒性。其高效的血脑屏障穿透能力使其在中枢镇痛领域具有显著优势。
随着对去甲氧基麻醉椒素药理作用机制的深入理解,其在镇痛治疗领域的临床应用前景逐渐明朗。相比传统阿片类镇痛药,去甲氧基麻醉椒素具有多靶点作用特性,可能降低依赖性及耐药性风险,减少副作用,满足临床对安全有效镇痛药物的迫切需求。
未来临床研究可重点关注其在慢性疼痛、神经性疼痛及癌痛等难治性疼痛中的应用潜力。此外,结合现代药物递送系统优化其生物利用度和靶向性,将进一步提升其临床价值。
除镇痛外,去甲氧基麻醉椒素的抗炎及神经保护作用也为其拓展至神经退行性疾病、炎症性疾病等领域提供了可能。多靶点、多功能的药理特性使其成为天然产物药物开发的典范。
然而,目前去甲氧基麻醉椒素的临床研究仍处于起步阶段,亟需系统的临床前安全性评价和临床试验验证,以推动其向临床应用转化。
去甲氧基麻醉椒素作为一种具有独特化学结构和多靶点镇痛机制的天然产物,展现出广阔的药理活性和良好的成药性。其在镇痛及相关疾病治疗中的潜力为天然产物药理学研究提供了新的方向。
未来研究应聚焦于其作用机制的深度解析、药代动力学的优化以及临床前安全性和有效性的系统评估。通过多学科交叉合作,去甲氧基麻醉椒素有望成为新一代安全高效的镇痛药物,为临床疼痛管理带来革新。
综上所述,去甲氧基麻醉椒素不仅丰富了天然产物的药理学知识体系,也为天然药物的现代化开发提供了宝贵的研究范例,值得持续关注和深入挖掘。

版权所有:© 成都普瑞法科技开发有限公司(2015)备案号:蜀ICP备15035167号-1 客服热线:400-829-7929
技术支持:南京库价















