
产品名称: 桑黄酚A
英文名: Hispolon
中文别名:
英文别名: Hispolone
Cas 号: 173933-40-9
产品编码:BP4816
分子式: C12H12O4
分子量: 220.224
来源: 桑黄
化合物类型: 酚类(Phenols)
纯度: 95%~99%
分析方法: HPLC-DAD or/and HPLC-ELSD
鉴定方法: 质谱(Mass), 核磁(NMR)
包装: 棕色小玻璃瓶,按客户需求包装。
存储: 贮存在避光密闭容器中,冷藏或者冷冻长期保存。
样品溶液最好临用新配。如果需要提前配制的话,最好分成独立包装冷冻保存(-20℃以下),临用前再取出解冻,通常可以保存2周。
可以满足克级以上大量需求,详情请咨询。
提供相关图谱和分析方法指导,可以提供包括色谱柱,标准品和分析方法在内的含量测定整体服务包,价格优惠。
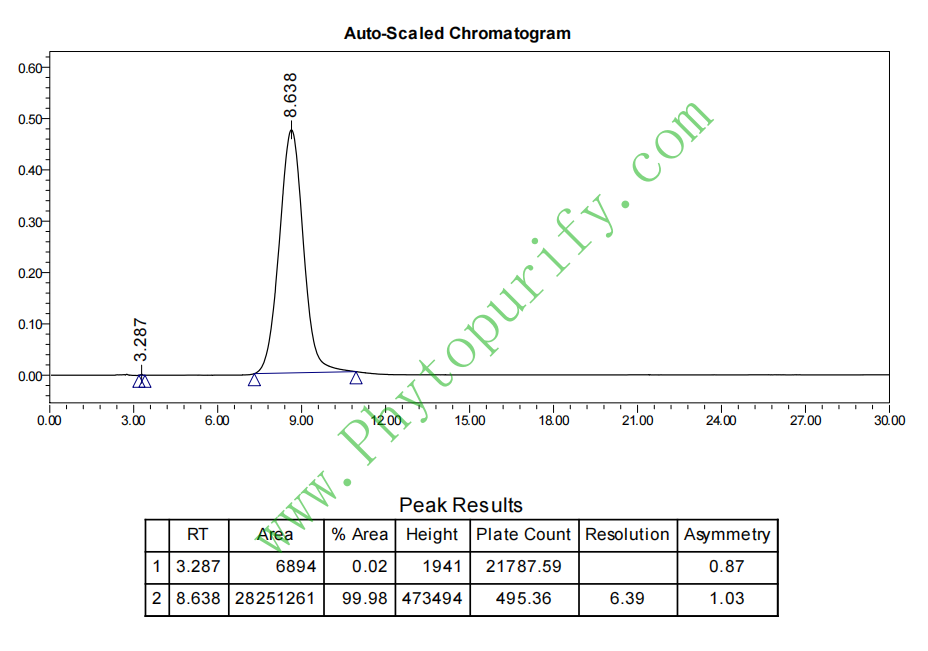
桑黄酚A的HPLC图谱
储存条件:短期保存 2~8℃,长期保存 -20 ~ -80℃
77.76
1.71
1.50
0.83
4.62
8.01
低
79.80
2.61
是
是
是
否
有
无
0.0
是
是
是
是

天然产物作为药物发现的重要源泉,在人类与疾病的漫长斗争史中扮演着无可替代的角色。真菌,尤其是大型药用真菌,因其独特的次生代谢产物库而备受关注。桑黄(Phellinus linteus),作为一种珍稀的药用真菌,在东亚地区(中国、日本、韩国)有着悠久的药用历史,传统上被用于治疗各种疾病,包括胃肠功能紊乱、炎症、肿瘤及增强免疫力。现代药理学研究证实,桑黄富含多糖、黄酮、吡喃酮、甾醇及酚类等多种生物活性成分,其中,桑黄酚A(Hispolon)作为其标志性的活性小分子酚类化合物,近年来引起了国内外学者的广泛研究兴趣。
桑黄酚A(Hispolon),化学名为6-(3,4-二羟基苯基)-4-羟基-2H-吡喃-2-酮,是一种具有独特苯乙烯基吡喃酮骨架的多酚类化合物。自1990年代首次从桑黄中分离鉴定以来,Hispolon被证实具有广泛而显著的药理活性,涵盖抗肿瘤、抗炎、抗氧化、抗糖尿病、抗病毒及肝保护等多个领域。其多靶点的作用特征,特别是对肿瘤发生发展中关键信号通路的调控,使其成为极具开发潜力的天然先导化合物。本文旨在系统综述Hispolon的化学结构、植物来源、药理活性、作用机制、成药性特征及临床应用前景,以期为该化合物的深入研究和开发利用提供全面的科学依据。
桑黄酚A(Hispolon)的化学结构是其生物活性的基础。其化学名为6-(3,4-二羟基苯基)-4-羟基-2H-吡喃-2-酮,分子式为C₁₁H₈O₅,分子量为220.2240 g/mol。从结构上看,Hispolon属于苯乙烯基吡喃酮类化合物,其核心骨架由一个2-吡喃酮(α-吡喃酮)环与一个邻苯二酚(儿茶酚)基团通过乙烯基桥连而成。具体而言,吡喃酮环的C-4位连接一个羟基(-OH),C-6位则通过一个乙烯基(-CH=CH-)与一个3,4-二羟基苯基相连。这种独特的结构赋予了Hispolon多种化学性质:邻苯二酚基团使其具有强大的抗氧化和金属螯合能力;α,β-不饱和酮结构(存在于吡喃酮环中)使其成为潜在的迈克尔加成受体,能够与生物大分子中的亲核基团(如半胱氨酸巯基)发生共价结合,这可能是其调控多种信号蛋白活性的关键机制之一。
在理化性质方面,Hispolon表现出典型的类药性特征。其脂水分配系数(LogP)为1.7068,表明其具有适中的亲脂性,有利于跨膜转运和与靶蛋白的疏水口袋结合。其拓扑极性表面积(TPSA)为77.7600 Ų,这一数值低于口服药物通常建议的上限(140 Ų),提示其具有良好的口服吸收潜力。水溶性参数为0.8279 mg/mL,属于微溶范畴,这在一定程度上可能影响其生物利用度,但通过合适的制剂技术(如纳米载体、环糊精包合等)有望得到改善。值得注意的是,计算机模拟预测显示,Hispolon透过血脑屏障(BBB)的能力较低,这提示其在中枢神经系统疾病治疗中的应用可能受限,但也意味着其在外周系统发挥作用时,中枢神经系统的副作用风险相对较低。此外,hERG抑制预测为“否”,Ames试验预测值为0.0,初步表明Hispolon在心脏毒性和遗传毒性方面具有较低的风险,为其作为候选药物的安全性提供了有利的早期证据。
Hispolon最初是从担子菌门、锈革孔菌科的真菌——桑黄(Phellinus linteus)的子实体或菌丝体中分离得到的。桑黄是一种寄生于桑树(Morus alba)及其他阔叶树上的多年生木腐菌,其子实体呈马蹄形或半球形,表面有龟裂,颜色从黄褐色至深褐色。除了P. linteus,Hispolon也被报道存在于其他Phellinus属真菌(如P. igniarius, P. baumii)以及少数其他真菌(如Inonotus属)中,但桑黄仍是其主要且最经典的来源。由于野生桑黄资源稀缺且生长周期长,目前人工栽培的桑黄菌丝体和子实体已成为获取Hispolon的主要来源。
Hispolon的提取通常依赖于经典的天然产物化学方法。由于Hispolon为中等极性的多酚类化合物,常用的提取溶剂包括甲醇、乙醇、乙酸乙酯及其水溶液。为了提高提取效率,现代提取技术如超声辅助提取、微波辅助提取、酶辅助提取等也被广泛应用。典型的提取流程如下:将干燥粉碎的桑黄子实体或菌丝体粉末,用一定浓度的乙醇(如70%-95%乙醇)在室温或加热条件下反复浸泡或渗漉提取。合并提取液,减压浓缩得到浸膏。随后,将浸膏分散于水中,依次用石油醚、乙酸乙酯、正丁醇进行液-液萃取。由于Hispolon的极性特征,其通常富集于乙酸乙酯萃取部位。该部位再经过多种色谱分离技术进行纯化,包括硅胶柱层析、ODS(十八烷基硅烷键合硅胶)反相柱层析、Sephadex LH-20凝胶柱层析以及制备型高效液相色谱(Prep-HPLC)。通过核磁共振波谱(NMR)和质谱(MS)等波谱学手段对纯化后的化合物进行结构鉴定,最终确证为Hispolon。近年来,随着绿色化学理念的推广,一些更环保、高效的提取方法,如深共晶溶剂提取、超临界流体萃取等,也开始被探索应用于Hispolon的提取,旨在提高产率并减少有机溶剂的使用。
Hispolon的药理活性谱非常广泛,涵盖了多个疾病领域,其中以抗肿瘤活性最为突出,研究也最为深入。
1. 抗肿瘤活性
Hispolon对多种人类癌细胞系表现出显著的增殖抑制作用,包括乳腺癌、前列腺癌、肺癌、肝癌、结直肠癌、胃癌、黑色素瘤、白血病和骨肉瘤等。其作用机制复杂,涉及诱导细胞凋亡、阻滞细胞周期、抑制肿瘤细胞迁移和侵袭、以及逆转多药耐药等多个方面。例如,在乳腺癌细胞中,Hispolon能够通过抑制STAT3信号通路,下调抗凋亡蛋白MCL1和BCL2的表达,从而诱导细胞凋亡。在前列腺癌中,它通过抑制雄激素受体信号和激活凋亡通路发挥抗癌作用。此外,Hispolon还被发现能够抑制DNA拓扑异构酶I和II(TOP1/TOP2A)的活性,干扰DNA复制和转录,这可能是其细胞毒性作用的重要机制之一。
2. 抗氧化活性
Hispolon分子结构中的邻苯二酚基团是其强大的自由基清除能力的结构基础。大量体外实验表明,Hispolon能够有效清除多种自由基,如DPPH自由基、ABTS阳离子自由基、羟基自由基和超氧阴离子自由基。它还能显著提高细胞内源性抗氧化酶(如超氧化物歧化酶SOD、过氧化氢酶CAT、谷胱甘肽过氧化物酶GPx)的活性,并增加还原型谷胱甘肽(GSH)的水平,从而保护细胞免受氧化应激损伤。这种抗氧化活性与其抗炎、肝保护及抗衰老等作用密切相关。
3. 抗炎活性
Hispolon在多种炎症模型中展现出显著的抗炎效果。它能够抑制脂多糖(LPS)刺激的巨噬细胞中一氧化氮(NO)、前列腺素E2(PGE2)以及促炎细胞因子(如TNF-α、IL-1β、IL-6)的产生。其机制主要涉及对核因子κB(NF-κB)和丝裂原活化蛋白激酶(MAPK,如MAPK1/ERK)信号通路的抑制。通过阻断这些关键炎症信号通路的激活,Hispolon能够从转录水平下调多种炎症介质的表达。
4. 抗糖尿病活性
Hispolon在糖尿病及其并发症的治疗中也显示出潜力。研究表明,Hispolon能够通过激活AMP活化蛋白激酶(AMPK)信号通路,促进葡萄糖摄取,改善胰岛素抵抗。在链脲佐菌素(STZ)诱导的糖尿病大鼠模型中,Hispolon给药能够显著降低血糖水平,改善血脂异常,并减轻糖尿病引起的肾脏和肝脏损伤。其抗糖尿病作用可能与其抗氧化和抗炎活性协同发挥。
5. 肝保护活性
Hispolon对多种化学性肝损伤(如四氯化碳CCl₄、对乙酰氨基酚APAP诱导)具有保护作用。它能够显著降低血清转氨酶(ALT、AST)水平,减轻肝组织坏死和炎症浸润。其肝保护机制与增强肝脏抗氧化防御能力、抑制肝星状细胞活化、以及调节凋亡相关蛋白(如Bax、Bcl-2)的表达有关。
6. 抗病毒活性
Hispolon还被报道具有抗病毒活性,包括对流感病毒、人类免疫缺陷病毒(HIV)和单纯疱疹病毒(HSV)等的抑制作用。其抗病毒机制可能涉及直接灭活病毒颗粒、抑制病毒吸附和进入宿主细胞、以及干扰病毒基因组的复制和转录。
Hispolon的药理活性并非源于单一靶点,而是通过多靶点、多通路协同作用实现的。其作用机制的核心可归纳为以下几个方面:
1. 调控细胞凋亡与存活信号通路
Hispolon最受关注的作用机制是其对凋亡信号通路的调控。它能够同时作用于内源性(线粒体)和外源性(死亡受体)凋亡途径。
* 抑制STAT3信号通路:Hispolon能够抑制STAT3的磷酸化和核转位,从而下调其下游靶基因,包括抗凋亡蛋白MCL1和BCL2。MCL1和BCL2的高表达是多种肿瘤细胞逃避凋亡的关键机制,Hispolon对其的抑制直接促进了肿瘤细胞的凋亡。
* 调节BCL2家族蛋白:除了抑制MCL1和BCL2,Hispolon还能上调促凋亡蛋白Bax的表达,并促进Bax从细胞质转移到线粒体膜,导致线粒体膜电位下降,释放细胞色素c,进而激活Caspase级联反应(Caspase-9, Caspase-3),最终诱导细胞凋亡。
* 激活MAPK通路:Hispolon对MAPK通路(如MAPK1/ERK, p38, JNK)的调控具有细胞和情境依赖性。在某些癌细胞中,它能够持续激活p38和JNK,这些应激激酶的活化通常与促凋亡信号相关。
2. 抑制肿瘤侵袭与转移
Hispolon能够有效抑制肿瘤细胞的迁移和侵袭能力,这对于控制肿瘤转移至关重要。
* 抑制基质金属蛋白酶(MMPs):Hispolon能够显著抑制MMP2和MMP9的表达和活性。MMPs是降解细胞外基质的关键酶,其活性增强是肿瘤细胞侵袭和转移的先决条件。Hispolon通过抑制MMPs,阻断了肿瘤细胞突破基底膜和向周围组织扩散的过程。
* 抑制HIF-1α:在缺氧条件下,HIF1A(HIF-1α)稳定表达并促进血管生成相关基因(如VEGF)的转录。Hispolon被发现能够抑制HIF-1α的蛋白积累和转录活性,从而抑制肿瘤新生血管的形成,切断肿瘤的营养供应,间接抑制肿瘤生长和转移。
3. 干扰细胞周期与DNA复制
Hispolon能够将肿瘤细胞周期阻滞在G1期或G2/M期,从而抑制细胞增殖。
* 抑制拓扑异构酶:Hispolon被证实是DNA拓扑异构酶I(TOP1)和II(TOP2A)的抑制剂。拓扑异构酶在DNA复制、转录和染色体分离中起关键作用。Hispolon通过稳定拓扑异构酶-DNA可裂解复合物,导致DNA双链断裂,从而触发细胞周期检查点,导致周期阻滞和细胞死亡。这一机制与许多临床抗癌药物(如喜树碱、依托泊苷)相似。
* 调控细胞周期蛋白:Hispolon能够下调细胞周期蛋白D1(Cyclin D1)和细胞周期蛋白依赖性激酶(CDK4/6)的表达,同时上调CDK抑制剂p21和p27的水平,共同导致细胞周期停滞。
4. 抗炎与抗氧化机制的交织
Hispolon的抗炎和抗氧化活性与其抗癌、肝保护等作用密切相关。
* 抑制NF-κB通路:Hispolon能够抑制IκBα的磷酸化和降解,从而阻止NF-κB p65亚基的核转位,抑制其转录活性。NF-κB是调控炎症反应和细胞存活的关键转录因子,其下游靶基因包括多种促炎细胞因子(TNF-α, IL-6)和抗凋亡蛋白。
* 激活Nrf2/ARE通路:Hispolon的邻苯二酚结构可以作为亲电体,激活核因子E2相关因子2(Nrf2)。Nrf2进入细胞核后与抗氧化反应元件(ARE)结合,启动一系列抗氧化酶(如SOD, CAT, HO-1)和解毒酶(如GST, NQO1)的转录,从而增强细胞的抗氧化防御能力。这一机制是其肝保护和细胞保护作用的重要基础。
此外,Hispolon还被发现能够通过调节雌激素受体α(ESR1)和芳香化酶(CYP19A1)的活性,在激素依赖性肿瘤(如乳腺癌)中发挥作用。这些多样化的分子靶点和信号通路共同构成了Hispolon复杂的药理作用网络。
将Hispolon从天然产物推向临床候选药物,需要对其成药性进行系统评价。如前所述,Hispolon在理化性质和初步毒理学预测上表现出一定的优势:分子量小(220 Da)、LogP适中、无hERG抑制风险、无Ames致突变性。这些特征符合“类药五规则”的大部分标准,为其口服给药提供了可能性。
然而,Hispolon的药代动力学特性是其成药性面临的主要挑战。目前关于Hispolon体内药代动力学的研究相对有限,但已有研究表明其口服生物利用度可能较低。这主要归因于以下几点:
1. 首过效应:作为多酚类化合物,Hispolon在肠道和肝脏中容易发生广泛的II相代谢,主要是葡萄糖醛酸化和硫酸化结合反应。这些代谢过程会显著降低其进入体循环的原型药物浓度。
2. 水溶性:尽管其水溶性(0.8279 mg/mL)尚可,但相对于理想的静脉注射或高口服剂量要求,仍显不足,可能限制其吸收。
3. 代谢稳定性:其分子中的酚羟基和α,β-不饱和酮结构是代谢酶(如细胞色素P450酶、儿茶酚-O-甲基转移酶COMT)的潜在作用位点,可能导致其在体内快速代谢清除。
针对这些挑战,提高Hispolon生物利用度的策略正在探索中。例如,将其制备成纳米脂质体、聚合物胶束或磷脂复合物,可以改善其溶解性和膜通透性,并保护其免受胃肠道降解。此外,设计Hispolon的前药,如将其酚羟基进行乙酰化或磷酸化修饰,可以暂时“封闭”其代谢位点,待其在体内被酶解后释放出原型药物,从而提高口服吸收。静脉注射给药途径可以完全规避首过效应,是另一种可行的选择,尤其适用于急性或重症治疗场景。
基于Hispolon丰富而确切的药理活性,其在临床转化方面展现出广阔的前景,主要集中于以下几个方向:
1. 抗肿瘤辅助治疗
Hispolon作为一种多靶点的天然抗癌化合物,其最大的优势在于能够同时作用于肿瘤细胞增殖、凋亡、侵袭、转移和血管生成等多个环节。它有可能作为一种化疗增敏剂或减毒剂,与常规化疗药物(如顺铂、阿霉素、紫杉醇)联合使用。例如,Hispolon通过抑制STAT3和NF-κB通路,可以逆转肿瘤细胞对某些化疗药物的耐药性。同时,其抗氧化和肝保护活性可能有助于减轻化疗引起的肝毒性、肾毒性等副作用。针对特定靶点(如MCL1、TOP1、HIF-1α)的抑制剂开发是当前抗癌药物研发的热点,Hispolon作为这些靶点的天然抑制剂,其结构可作为先导化合物,通过药物化学修饰(如结构简化、优化药效团)来开发具有更高选择性和更强活性的衍生物。
2. 代谢性疾病与慢性炎症管理
Hispolon的抗糖尿病、抗氧化和抗炎活性使其在治疗2型糖尿病及其并发症(如糖尿病肾病、非酒精性脂肪性肝病NAFLD)方面具有潜力。作为一种天然的AMPK激活剂和Nrf2诱导剂,Hispolon或其衍生物有望开发成一种改善胰岛素抵抗、调节脂质代谢和减轻氧化应激的膳食补充剂或功能性食品成分。在慢性炎症性疾病(如类风湿性关节炎、炎症性肠病)中,其通过抑制NF-κB和MAPK通路的抗炎作用也值得进一步探索。
3. 肝脏保护剂
鉴于其显著的肝保护活性,Hispolon有潜力被开发用于预防和治疗各种原因引起的肝损伤,包括药物性肝损伤、酒精性肝病和病毒性肝炎。其多靶点的保护机制(抗氧化、抗炎、抗凋亡)使其在应对复杂的肝脏病理过程中可能比单一靶点的药物更具优势。
尽管前景光明,但Hispolon的临床转化仍面临诸多挑战。首先,其体内药代动力学特性不佳是最大的瓶颈,需要依靠先进的制剂技术或前药设计来突破。其次,目前绝大多数研究仍停留在体外细胞和动物模型水平,缺乏高质量的人体临床试验数据来验证其有效性和安全性。此外,Hispolon的多靶点特性虽然带来了广泛活性,但也可能增加脱靶效应的风险,需要对其长期用药的安全性进行更全面的评估。
桑黄酚A(Hispolon)作为来自药用真菌桑黄的一种天然多酚化合物,凭借其独特的苯乙烯基吡喃酮结构和多靶点的药理作用特征,在抗肿瘤、抗炎、抗氧化、抗糖尿病及肝保护等多个疾病领域展现出令人瞩目的潜力。其作用机制涉及对STAT3、NF-κB、MAPK、Nrf2、拓扑异构酶、MMPs及BCL2家族蛋白等多个关键信号分子和靶点的调控,体现了天然产物“多靶点、多通路”协同作用的优势。初步的成药性评价显示其具有较好的类药性基础,但口服生物利用度低是其临床转化面临的核心障碍。
未来的研究应聚焦于以下几个关键方向:一是利用药物化学手段,以Hispolon为先导,设计合成具有更高代谢稳定性和生物利用度的衍生物;二是开发高效的药物递送系统,如纳米制剂,以改善其药代动力学特性;三是开展更深入的体内药效学研究,特别是在与现有药物联合应用的协同效应和减毒作用方面;四是推动其进入规范的临床前毒理学评价和临床试验阶段。随着研究的不断深入和技术的进步,Hispolon及其衍生物有望成为治疗人类重大疾病的新一代候选药物,为天然产物药物的开发提供又一成功范例。

版权所有:© 成都普瑞法科技开发有限公司(2015)备案号:蜀ICP备15035167号-1 客服热线:400-829-7929
技术支持:南京库价















