
产品名称: 马兜铃酸C
英文名: Aristolochic acid C
中文别名:
英文别名: Aristolochic acid C
Cas 号: 4849-90-5
产品编码:BPF0516
分子式: C16H9NO7
分子量: 327.248
来源:
物理性状:
化合物类型: Alkaloids
纯度: 95%~99%
分析方法: HPLC-DAD or/and HPLC-ELSD
鉴定方法: 质谱(Mass), 核磁(NMR)
包装: 棕色小玻璃瓶,按客户需求包装。
可以满足克级以上大量需求,详情请咨询。
提供相关图谱和分析方法指导,可以提供包括色谱柱,标准品和分析方法在内的含量测定整体服务包,价格优惠。
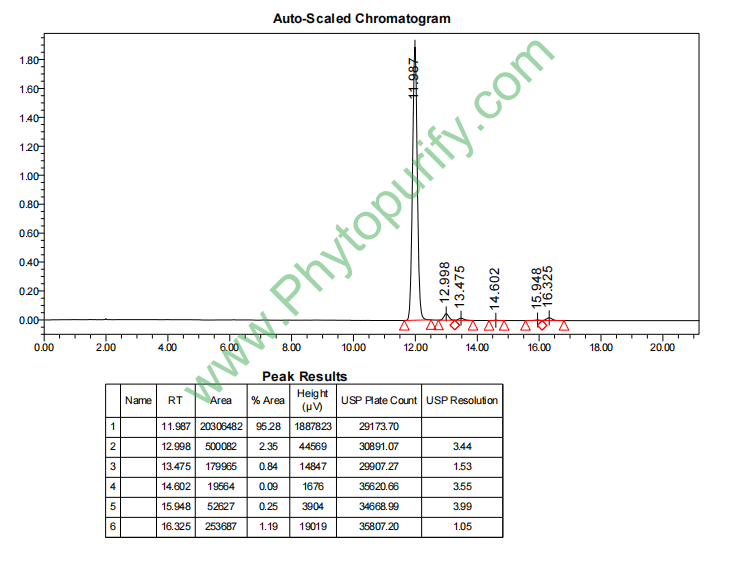
马兜铃酸C的HPLC图谱
储存条件:短期保存 2~8℃,长期保存 -20 ~ -80℃
119.13
3.23
0.14
0.17
1.81
18.66
低
91.22
2.66
是
否
否
否
有
有
2.4
是
是
是
是

马兜铃酸类化合物(Aristolochic acids, AAs)是一类天然存在于马兜铃属(Aristolochia)和细辛属(Asarum)植物中的硝基菲羧酸衍生物,因其显著的肾毒性和致癌性而备受关注。自20世纪90年代“马兜铃酸肾病”(Aristolochic Acid Nephropathy, AAN)被首次报道以来,这类化合物已成为天然产物毒理学研究的热点。马兜铃酸C(Aristolochic acid C, AAC)作为马兜铃酸家族的重要成员之一,其化学结构、生物活性及毒性机制既与马兜铃酸I(AAI)和马兜铃酸II(AAII)存在共性,又展现出独特的分子特征。
马兜铃酸C的CAS号为4849-90-5,分子式为C₁₇H₁₃NO₆,属于硝基菲类化合物。与AAI相比,AAC在菲环的C-8位缺少一个甲氧基,而保留了一个羟基,这一结构差异不仅影响了其理化性质,也对其与生物大分子的相互作用模式产生了深远影响。早期研究主要关注AAI和AAII的毒性,但随着对马兜铃酸代谢组学和毒理学的深入探索,AAC在肾毒性、氧化应激及细胞凋亡调控中的独特作用逐渐被揭示。
值得注意的是,马兜铃酸C不仅是磷脂酶A2(PLA2)的抑制剂,还能破坏拟南芥间期细胞的周质微管列阵并抑制根生长,提示其可能通过干扰细胞骨架动力学发挥生物学效应。在哺乳动物系统中,AAC的肾毒性涉及BCL2家族蛋白、NFE2L2抗氧化通路、TP53肿瘤抑制因子以及CASP3介导的凋亡级联反应,这些分子事件共同构成了马兜铃酸肾病复杂的病理网络。
本文将从化学结构、植物来源、药理活性、分子机制、成药性评价及临床应用前景等角度,对马兜铃酸C的研究进展进行系统综述,旨在为天然产物毒理学和药物安全性评估提供参考。
马兜铃酸C的分子式为C₁₇H₁₃NO₆,分子量为327.2480 g/mol。其核心结构为菲环骨架,在菲环的C-1位连接羧基(-COOH),C-4位连接硝基(-NO₂),C-8位为羟基(-OH),C-10位为甲氧基(-OCH₃)。与马兜铃酸I(AAI,C₁₇H₁₁NO₇,C-8位为甲氧基)相比,AAC在C-8位以羟基取代了甲氧基,这一结构差异导致其极性和氢键供体能力增强。与马兜铃酸II(AAII,C₁₆H₉NO₆,C-8位为氢原子)相比,AAC的C-8位羟基使其具有更强的亲水性和反应活性。
根据计算化学预测及实验测定数据,马兜铃酸C的理化性质参数如下:
马兜铃酸C的紫外-可见吸收光谱在250-400 nm范围内呈现特征性吸收峰,其中约260 nm和320 nm处的强吸收归因于菲环的π→π跃迁,而约390 nm处的弱吸收可能与硝基的n→π跃迁有关。红外光谱中,约1700 cm⁻¹的强吸收峰对应羧基的C=O伸缩振动,约1520 cm⁻¹和1350 cm⁻¹的吸收峰分别对应硝基的不对称和对称伸缩振动。核磁共振氢谱中,菲环上的芳香质子信号分布在δ 7.0-8.5 ppm区域,其中C-8位羟基的质子信号出现在δ 9.5-10.5 ppm,且易与溶剂发生交换。
马兜铃酸C主要存在于马兜铃属(Aristolochia)和细辛属(Asarum)植物中,这些植物在传统中医药、阿育吠陀医学及南美民间医药中有着悠久的应用历史。以下为AAC含量较高的代表性植物:
马兜铃酸C的提取通常采用有机溶剂萃取结合色谱分离的方法,具体流程如下:
1. 原料预处理:将干燥的植物材料粉碎至40-60目,用石油醚或正己烷脱脂,去除叶绿素和脂溶性杂质。
2. 溶剂提取:采用甲醇或70%乙醇水溶液作为提取溶剂,料液比1:10-1:20(w/v),在60-80℃下回流提取2-3次,每次2-4小时。超声辅助提取(40-60 kHz,30-60分钟)可提高提取效率约20-30%。
3. 液-液萃取:将提取液浓缩后,用乙酸乙酯或正丁醇进行液-液萃取,马兜铃酸类化合物主要分配至乙酸乙酯相。调节pH至2-3(使用稀盐酸),可促进AAC以游离酸形式进入有机相。
4. 柱色谱分离:采用硅胶柱色谱(200-300目),以氯仿-甲醇-乙酸(90:10:1至70:30:1)梯度洗脱,收集含AAC的流分。进一步通过Sephadex LH-20凝胶柱色谱(甲醇洗脱)或制备型高效液相色谱(C18反相柱,乙腈-水-甲酸体系)纯化,可获得纯度>98%的AAC单体。
5. 质量控制:采用高效液相色谱-二极管阵列检测(HPLC-DAD)或液相色谱-质谱联用(LC-MS/MS)进行定量分析,检测波长通常设定为250 nm或320 nm。AAC的保留时间在C18柱上通常介于AAII和AAI之间,这与羟基取代基的极性效应一致。
马兜铃酸C的肾毒性是其最受关注的药理活性,也是限制其药用价值的主要障碍。体内外实验均证实AAC可诱导肾小管上皮细胞损伤,其毒性强度约为AAI的1/5-1/3,但高于AAII。
细胞水平研究:在人肾近端小管上皮细胞(HK-2细胞)中,AAC(10-100 μM)处理24-48小时可导致细胞活力呈浓度依赖性下降,半数抑制浓度(IC₅₀)约为40-60 μM。形态学观察显示,AAC处理后的细胞出现核浓缩、凋亡小体形成及线粒体肿胀等典型凋亡特征。流式细胞术检测表明,AAC可诱导细胞周期阻滞于G0/G1期,并增加亚二倍体细胞比例。
动物模型研究:在C57BL/6小鼠中,腹腔注射AAC(5-20 mg/kg/d,连续7天)可导致血尿素氮(BUN)和血清肌酐(SCr)水平显著升高,肾组织病理学检查显示肾小管上皮细胞变性、坏死及间质纤维化。值得注意的是,AAC的肾毒性存在明显的性别差异,雄性小鼠的肾损伤程度重于雌性,这可能与雄激素对代谢酶活性的调节有关。
马兜铃酸C是磷脂酶A2(PLA2)的竞争性抑制剂,其IC₅₀约为15-25 μM。PLA2催化膜磷脂sn-2位脂肪酸的水解,释放花生四烯酸,是炎症反应的关键酶。AAC通过其菲环骨架与PLA2的疏水通道结合,而硝基和羧基则与活性位点的组氨酸和天冬氨酸残基形成氢键。这一抑制活性可能部分解释了马兜铃酸类化合物的抗炎作用,但同时也可能通过干扰膜磷脂代谢加重细胞损伤。
在拟南芥(Arabidopsis thaliana)根尖细胞中,AAC(10-50 μM)处理可导致间期细胞的周质微管列阵发生紊乱,表现为微管束的断裂、解聚和异常聚集。这一效应与微管蛋白的直接结合无关,而是通过激活微管相关蛋白的磷酸化信号通路实现。AAC对植物微管的破坏作用与其对动物细胞微管的影响存在差异,提示其可能具有物种选择性的细胞骨架毒性。
马兜铃酸C的肾毒性涉及多条信号通路的交叉调控,其中BCL2家族蛋白、NFE2L2抗氧化通路、TP53肿瘤抑制因子及CASP3介导的凋亡级联反应构成了核心机制网络。
1. BCL2家族与线粒体凋亡通路
AAC处理可导致肾小管上皮细胞中抗凋亡蛋白BCL2的表达下调,而促凋亡蛋白BAX的表达上调,BAX/BCL2比值显著升高。这一变化促使BAX从细胞质转位至线粒体外膜,形成寡聚体通道,导致线粒体膜电位(ΔΨm)下降、细胞色素c释放至胞质,进而激活CASP9和CASP3,最终执行凋亡程序。免疫荧光染色显示,AAC处理后的HK-2细胞中,BAX的线粒体定位增加约3-5倍,而BCL2的线粒体保护作用减弱。
2. NFE2L2抗氧化通路
NFE2L2(NRF2)是细胞抗氧化防御的主转录因子。AAC可诱导NFE2L2从KEAP1复合物中解离并核转位,上调下游抗氧化酶如血红素加氧酶-1(HO-1)、NAD(P)H醌氧化还原酶1(NQO1)及谷胱甘肽S-转移酶(GST)的表达。然而,这种适应性抗氧化反应在持续暴露下逐渐耗竭,导致活性氧(ROS)积累。值得注意的是,AAC诱导的ROS主要来源于NADPH氧化酶(NOX2)的激活,而非线粒体电子传递链。NOX2抑制剂(如apocynin)可部分缓解AAC的细胞毒性,提示氧化应激在肾损伤中发挥关键作用。
3. TP53信号通路
AAC可激活TP53(p53)蛋白,表现为p53的磷酸化(Ser15位点)和乙酰化水平升高,以及其转录靶基因CDKN1A(p21)和BAX的上调。p21的诱导导致细胞周期阻滞于G1/S检查点,为DNA损伤修复争取时间。然而,当DNA损伤超出修复能力时,p53转而促进凋亡。在p53敲除的HK-2细胞中,AAC的细胞毒性显著降低,表明p53是AAC肾毒性的关键中介因子。
4. 肾损伤标志物
AAC处理可诱导肾损伤分子-1(KIM-1/HAVCR1)和中性粒细胞明胶酶相关脂质运载蛋白(NGAL/LCN2)的表达显著上调。KIM-1是一种跨膜糖蛋白,在受损肾小管上皮细胞中高表达,可作为早期肾损伤的生物标志物。NGAL则参与铁离子转运和炎症反应,其尿中排泄量与肾小管损伤程度呈正相关。在AAC处理的小鼠模型中,尿KIM-1和NGAL水平在给药后24小时内即升高,早于血肌酐和尿素氮的变化。
马兜铃酸C的硝基在细胞内经还原酶(如NAD(P)H醌氧化还原酶、细胞色素P450还原酶)代谢后,形成N-羟基马兜铃酸内酰胺,后者进一步与DNA中的腺嘌呤和鸟嘌呤碱基形成共价加合物。AAC-DNA加合物的主要类型为7-(脱氧腺苷-N⁶-基)马兜铃酸内酰胺(dA-AAI)和7-(脱氧鸟苷-N²-基)马兜铃酸内酰胺(dG-AAI)。这些加合物可导致DNA聚合酶读码错误,引起A→T颠换突变,这是马兜铃酸致癌性的分子基础。与AAI相比,AAC形成的DNA加合物量较少,但加合物在组织中的滞留时间更长,这可能与其C-8位羟基的代谢稳定性有关。
AAC对微管的影响并非通过与微管蛋白直接结合,而是通过激活RhoA/ROCK信号通路,导致微管相关蛋白MAP4的过度磷酸化。磷酸化的MAP4从微管表面解离,降低微管的稳定性,最终导致微管骨架的解聚。此外,AAC还可抑制微管正端追踪蛋白(如EB1)的定位,干扰微管与细胞皮层的相互作用,影响细胞极性和定向迁移。
基于Lipinski五规则和Veber规则,马兜铃酸C的成药性参数如下:
从上述参数看,AAC基本符合口服药物的理化性质要求,但TPSA偏高可能限制其肠道吸收。此外,Ames试验阳性结果提示其具有遗传毒性风险,这是成药性的最大障碍。
吸收:AAC的口服生物利用度较低(大鼠实验中约为15-25%),这可能与其在胃肠道的溶解度有限及P-糖蛋白(P-gp)的外排作用有关。Caco-2细胞单层实验显示,AAC的表观渗透系数(Papp)为1.2-2.5 × 10⁻⁶ cm/s,属于中等渗透性化合物。
分布:AAC在体内分布广泛,肾脏和肝脏中的浓度最高,这与肾毒性和肝毒性的靶器官一致。血浆蛋白结合率约为85-92%,主要与白蛋白结合。表观分布容积(Vd)约为0.5-1.0 L/kg,提示组织分布有限。
代谢:AAC的主要代谢途径包括:①硝基还原为N-羟基马兜铃酸内酰胺(由细胞色素P450 1A2和NAD(P)H醌氧化还原酶催化);②C-8位羟基的葡萄糖醛酸结合(由UGT1A1和UGT1A9催化);③菲环的氧化(由CYP1A2和CYP3A4催化)。代谢产物主要通过胆汁排泄,部分经肾小球滤过。
排泄:AAC及其代谢物的半衰期(t₁/₂)约为8-12小时,但DNA加合物的清除半衰期可达数周至数月。约60-70%的给药剂量在72小时内通过粪便排泄,20-30%通过尿液排泄。值得注意的是,AAC在肾脏中的蓄积是其长期肾毒性的重要原因。
基于现有数据,马兜铃酸C的毒性风险可归纳如下:
马兜铃酸C作为马兜铃属植物的活性成分之一,曾广泛存在于传统中药制剂中,如“龙胆泻肝丸”、“八正散”等。然而,自2003年以来,多个国家和地区已禁止或限制含马兜铃酸类化合物的中药材使用。尽管如此,由于植物鉴定错误、替代品使用不当及非法贸易等原因,马兜铃酸肾病仍在全球范围内时有发生。
尽管肾毒性限制了马兜铃酸C的直接药用价值,但其独特的分子结构为药物设计提供了启示:
结构修饰:通过化学修饰降低毒性,例如将硝基还原为氨基,或将羧基酯化,可能获得毒性较低的衍生物。已有研究报道,AAC的甲酯衍生物(AAC-methyl ester)的肾毒性降低约5倍,但仍保留PLA2抑制活性。
靶向递送:利用纳米载体(如脂质体、聚合物胶束)将AAC靶向递送至肿瘤组织,可降低全身暴露量。初步研究表明,AAC-脂质体在荷瘤小鼠中的肾毒性较游离药物降低约60%。
作为工具分子:AAC作为PLA2抑制剂和微管破坏剂,可用于研究炎症和细胞骨架动力学的分子机制。其独特的DNA加合物形成能力也使其成为研究化学致癌机制的理想模型化合物。
鉴于马兜铃酸C的遗传毒性和肾毒性,建议采取以下措施:
马兜铃酸C作为马兜铃酸家族的重要成员,其化学结构、药理活性及毒性机制既与AAI和AAII存在共性,又展现出独特的分子特征。C-8位羟基的引入使其极性和代谢途径发生改变,导致肾毒性强度低于AAI但高于AAII。AAC通过BCL2/BAX介导的线粒体凋亡通路、NFE2L2抗氧化通路、TP53信号通路及NOX2氧化应激途径,诱导肾小管上皮细胞损伤,其DNA加合物形成能力则构成遗传毒性基础。
尽管AAC的直接药用价值因肾毒性而受限,但其作为PLA2抑制剂和微管破坏剂的活性为药物设计提供了结构模板。通过化学修饰、靶向递送及毒性规避策略,有望开发出低毒性的衍生物。同时,AAC作为研究化学致癌和肾毒性机制的模型化合物,在基础毒理学研究中具有重要价值。
未来,随着代谢组学、表观遗传学及单细胞测序技术的发展,对马兜铃酸C毒性机制的认知将更加深入。建立基于风险评估的监管标准,加强中药材安全性评价,开发有效的解毒策略,是保障公众健康的关键举措。马兜铃酸C的研究历程警示我们,天然产物既是药物发现的宝库,也可能隐藏着潜在的健康风险,唯有通过严谨的科学评估,才能实现传统医药的现代化和可持续发展。

版权所有:© 成都普瑞法科技开发有限公司(2015)备案号:蜀ICP备15035167号-1 客服热线:400-829-7929
技术支持:南京库价















