2019-01-27西红花苷和西红花酸的药动学及制剂学研究进展
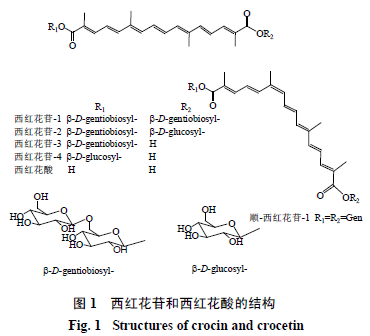
西红花苷(crocin)来源于鸢尾科植物西红花Crocus sativus Linne干燥暗红柱头及栀子Gardeniajasminoides Ellis干燥成熟果实,主要包括西红花苷-1、西红花苷-2、西红花苷-3和西红花苷-4等水溶性不饱和脂肪酸葡萄糖糖苷,其苷元为西红花酸(图1)。以西红花苷-1为代表的这类衍生物具有保护神经元 [1]、抗肿瘤[2]、抗血栓[3]、抗抑郁[4]、免疫调节[5]、降血压[6]等药理活性。同时,其苷元西红花酸(crocetin)也显示出抗食管癌[7]、改善溃疡性结肠炎[8]、保护神经元 [9]、抗动脉粥样硬化[10]、抗肿瘤[11]等药理活性。药动学研究包括药物在生物体内的吸收、分布、代谢及排泄机制,药动学研究有助于阐明药物作用机制、优化剂量方案、减少不良反应、指导临床合理用药,在中药研发中具有重要意义。近年来学者对西红花苷和西红花酸广泛开展了药动学研究,揭示其体内代谢规律。此外,西红花苷属高度不饱和类胡萝卜素,稳定性差,对高温、光线、低pH值敏感[12],其生物利用度也相对较低,故限制了其临床应用。西红花酸也由于其溶解性差[13],限制了其药理功效发挥。为进一步提高西红花苷和西红花酸生物利用度及稳定性,各国学者广泛开展了西红花苷和西红花酸脂质体、微囊、乳剂、纳米粒、软胶囊、缓释片等研究,以提高西红花苷和西红花酸药理功效。本文综述近年来西红花苷和藏红花酸药动学和新型递药系统研究进展,为进一步阐明西红花苷和西红花酸药理功效机制及该类天然产物的深入研究与开发利用提供思路和参考。

1 西红花苷和西红花酸体内药动学研究
同一药物给药途径不同,体内吸收、组织分布、代谢和排泄也不同。目前,研究报道西红花苷和西红花酸代谢研究的给药途径有iv、ig、im及ip。数据显示,西红花苷和西红花酸采用不同给药途径,其体内药动学特征差异较大。
1.1 吸收
药物吸收(absorption)指药物由给药部位进入血液循环过程。数据显示,西红花苷在肠道不易吸收,而是水解成西红花酸后吸收进入血液循环。
1.1.1 西红花苷动物体内吸收 金玉燕等[14]测定西红花苷-1油水分配系数(log P)为−1.03,一般认为logP值为2~3的药物在肠道中易吸收,log P<0时则极不易被肠道吸收[15],以上数据显示,西红花苷 I在肠道不易吸收。而杜鹏等采用在体小肠回流模型研究西红花苷-1肠道吸收,结果显示,西红花苷-1在十二指肠、空肠、回肠和结肠不吸收,且各肠段均降解10%左右。金玉燕等[14]研究也得出类似结论,并证实了减少部分与肠内酶有关。此外,Lautenschläger等[17]选择Caco-2细胞模型研究西红花苷-1小肠吸收情况,结果显示,西红花苷-1在基底侧浓度低于检测限。西红花苷-1表观渗透系数Papp(AP→BL)为(2.1±0.8)×10−7 cm/s,一般认为Papp>1×10−6药物吸收良好[18],进一步证实西红花苷不能透过肠道屏障。
此外,Karkoula等[19]以ip方式给予小鼠西红花苷-1,采用高灵敏度UPLC-PDA法检测血液及脑组织中西红花苷-1和西红花酸含量,结果显示,小鼠血液和脑组织中均未检测出西红花酸,只检测出西红花苷 I,此研究首次证明了西红花苷 I可穿透血脑屏障(BBB)。而李向阳等[20]研究藏药佐太对西红花苷-1在大鼠体内药动学影响,结果显示,连续ig佐太10 mg/ (kg∙d) 7 d和12 d后可增加西红花苷- 1的吸收,显著降低了清除率 [对照组为(0.019±0.005)L/(kg∙min),实验组为(0.011±0.005)L/(kg∙min),P<0.01]。此外,朱俊博等[21]研究结果显示,西红花苷-1经im后吸收较快,分布广泛,平均体内滞留时间短。
1.1.2 西红花酸动物体内吸收 刘同征等[22]研究显示,大鼠直接ig西红花酸,其体内药-时曲线符合口服给药二室模型。研究显示,西红花酸体内吸收和消除快,提示若西红花酸直接给药,则需考虑缓释制剂或者采用多次给药方式,延长其体内滞留和作用时间。杜鹏等[23]研究显示按不同剂量ig给予大鼠西红花酸,药-时曲线也符合口服给药二室模型。西红花酸体内达峰时间快,吸收迅速,呈一级动力学消除。上述研究表明,给药方式不同但研究结果相似,显示西红花酸在肠道水解速度快。而且,研究显示单次和多次ig大鼠西红花酸时,其达峰浓度相差不大 [单次为(0.83±0.31)μg/mL,多次为(0.79±0.25)μg/mL],进一步证实了西红花酸体内消除快,累积小,半衰期短[24]。
另一方面,张颖等[25]研究显示大鼠ig西红花苷-1后西红花酸血药浓度有2次达峰,且浓度波动不大。与直接ig西红花酸[23]相比,该研究表明大鼠ig相同剂量西红花苷-1,西红花酸表现出更高的峰浓度、更长的半衰期和平均滞留时间,显示出更为良好药动学特征。此外,Lautenschläger等[17]研究显示西红花酸能够以较快速度渗透Caco-2细胞,Papp(BL→AP)为(2.5±0.2)×10−5 cm/s,西红花酸可穿透肠道屏障且在肠道吸收良好。研究发现西红花酸主要通过被动跨细胞转运方式渗透肠道,无需特定载体。
1.1.3 西红花酸人体吸收 人体临床试验研究方面,Umigai等[26]研究健康人口服西红花酸的吸收情况,采用HPLC法检测西红花酸的血药浓度。10名健康受试者,男女各半,单次口服西红花酸7.5、15.0、22.5 mg,结果显示,给药后1 h血浆中可检测出西红花酸,药物在体内达峰时间(tmax)为4.0~4.8 h,随后逐渐降低,24 h时降低至定量限。最大血药浓度(Cmax)为100.9~279.7 ng/mL,药-时曲线下面积(AUC)为556.5~1 720.8 ng∙h/mL,生物半衰期(t1/2)为6.1~7.5 h,Cmax和AUC值与剂量呈正比,且呈剂量依赖性。相对其他类胡萝卜素[27],西红花酸具有更快吸收的特点。Chryssanthi等[28]采用HPLC法检测健康人饮用200 mg西红花茶后血浆中西红花酸的浓度。结果显示,10.7和18.6 min在血浆中分别检测出反式西红花酸和顺式西红花酸,24 h后血浆中还残留反式西红花酸。75%的人体内顺式西红花酸占总西红花酸的25%~50%,表明西红花酸在人体内发生了异构化,该研究首次发现西红花酸在体内发生结构异构化。此外,Hooshang等[29]研究显示健康志愿者单次口服16 mg西红花酸胶囊后西红花酸血药浓度范围为0.09~0.35 μg/mL。
1.2 分布
药物分布(distribution)指药物从给药部位吸收进入血液后,由循环系统运送至体内各脏器、组织、体液和细胞的转运过程。不同给药途径会影响药物分布速度。与其他类胡萝卜素(如β-胡萝卜素、叶黄素、番茄红素)相比,西红花酸亲水性相对较强,相对分子质量更小,故在体内器官组织分布广泛。杜鹏等[23]研究发现大鼠ig西红花酸后,其在体内分布很快达到平衡,肝脏、肺、肾、脾、子宫和卵巢l h达峰,而脂肪和睾丸则为2 h,各组织脏器中西红花酸的浓度依次为肝>肺>子宫≈卵巢>肾>脂肪>睾丸。钱之玉等[30]研究了大鼠iv西红花苷-1后在各组织中的分布情况,结果表明西红花苷-1在血、肺和肾组织中的浓度最高,肝、心、胃次之,而脑、脂肪和睾丸中西红花苷-1浓度低于检测限,表明西红花苷和西红花酸体内分布良好。此外,Lautenschläger等[17]选择猪脑毛细血管内皮细胞(BCEC)模型和血-脑脊液屏障细胞(BCSFB)模型研究西红花酸能否渗透BBB,结果显示,西红花酸在29 h内缓慢渗透BCEC和BCSFB,渗透BCEC的Papp为(1.5±0.1)×10−6cm/s,渗透BCSFB的Papp为(3.9±0.2)×10−6cm/s,表明西红花酸可穿透BBB,这可能是西红花酸能进入中枢神经系统发挥药效的机制。
1.3 代谢
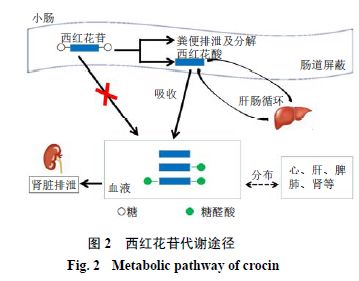
西红花苷-1糖苷键属羧酸酯键,而羧酸酯酶广泛分布于体内各组织器官中,小肠和肝脏都是其主要分布器官[31]。杜鹏等[16]研究显示西红花苷-1经ig给药,不能以原型吸收入血,而是经肠道酶或细菌酶作用水解成苷元(西红花酸)后吸收入血,且在血浆中有2次达峰。此外,Xi等[24]给予大鼠ig西红花苷-1,结果显示,1 h血浆中检测出少量西红花酸,而未检测出西红花苷-1。Asai等[32]研究则进一步显示,西红花酸可直接吸收进入血液,而西红花苷可能在消化道内(吸收前)或者肠黏膜(吸收过程中)水解成西红花酸后吸收入血,而西红花酸可能在肠黏膜和肝脏中部分代谢成西红花酸-单(双)葡萄糖醛酸缀合物,与西红花酸共同存在于血浆中。此外,冯晓宾等[33]给予大鼠ig西红花水提液,血浆中未检测出西红花苷,而检测出西红花酸和西红花酸单葡萄糖醛酸结合物。西红花酸在30 min出现最高血药浓度,120min出现第2个峰值,出现此现象的原因可能是产生了肝肠循环。这与张颖等[25]的研究结论一致,提示西红花苷的代谢物西红花酸在血液中会出现双峰现象,并且2次达峰浓度相近。
Zhang等[34]采用超高灵敏的UPLC-MS/MS法同时研究西红花苷和西红花酸在大鼠体内的药动学。ig相同剂量(30、60、120 μmol/kg)西红花苷- 1和西红花酸,前者大鼠血液中西红花酸Cmax和AUC值明显大于后者。
1.4 排泄
药物排泄(excretion)指吸收进入体内药物及其代谢产物从体内排出体外的过程。西红花苷排泄方式与给药方式密切相关。钱之玉等[30]研究显示大鼠iv4 mg/kg西红花苷-1,结果其1.92%和36.95%分别由粪和尿排泄出,仅0.74%由胆汁排泄出,12 h内药物基本排泄完全。杜鹏等[23]研究显示ig给予大鼠50 mg/kg西红花苷-1,24 h粪排泄率为(59.50±13.56)%,肠内滞留药量为(20.43±9.41)%,占给药剂量的80%,24 h尿中检测不出西红花苷-1。以上结果表明,iv西红花苷主要经肾脏排泄,ig主要经粪便排泄。
1.5 小结
西红花苷相对分子质量大,极性强,口服不能以原型药物透过肠道屏障,而是在肠道内先水解为相对分子质量较小、脂溶性较强的西红花酸,然后以被动跨细胞扩散的方式进入血液循环,分布于心、肝、脾、肺、肾、大脑、子宫、卵巢、脂肪、睾丸等组织。西红花苷和西红花酸具有广泛的药理活性,如心肌缺血保护[35]、肝保护[36]及脑损伤保护[37]等,而有关该药物相应组织器官的体内分布数据则印证这一系列药效。此外,西红花酸进入肠道后,部分西红花酸还可在小肠黏膜继续代谢为西红花酸-单(双)葡萄糖醛酸缀合物,与西红花酸共同存在于血液中,前者可能通过肾脏排泄,而口服未被肠道吸收的西红花苷则主要经粪便排泄。西红花苷和西红花酸传统主要以口服给药为主,药动学研究结果提示,西红花苷体内活性分子可能是西红花酸。同时,西红花苷和西红花酸具有较快的血浆清除速度,若要更好发挥西红花苷和西红花酸药效,维持体内有效血药浓度,可能需增加给药频率,或借助于现代递药系统研究西红花苷的新剂型。西红花苷和西红花酸代谢途径见图2。

2 西红花苷和西红花酸制剂学研究
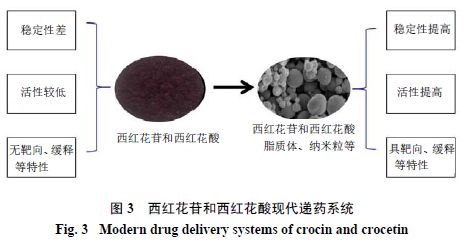
西红花苷和西红花酸药理活性广泛,但通过以上药动学研究显示,二者口服给药在体内生物利用度较低、生物半衰期短、消除快,尤其是西红花苷,口服吸收较差,并且在肠道内不稳定。因此,有必要研究西红花苷和西红花酸新型递药系统。近年来,学者利用现代递药新技术将西红花苷和西红花酸制备成脂质体、微囊、复乳、纳米粒、软胶囊及缓释片等,以增加二者的稳定性、改善溶解性、提高生物利用度并最终增强其药理活性。
2.1 脂质体
脂质体(liposome)是由脂质双分子层组成的内部为水相闭合囊泡。按脂质体结构类型可分为单层脂质体、多层脂质体和多囊脂质体;按结构性能又可分为普通脂质体和特殊性能脂质体[38]。纳米脂质体(nanoliposome)是一类小单室脂质体,可迅速并靶向进入靶部位。基于西红花苷具有显著的抗癌作用,Mousavi等[39]以二硬脂酰磷脂酰甘油(DSPG)、二硬脂酰磷脂酰胆碱(DSPC)和胆固醇(Chol)为原料,通过脱水和再水合法(DRV)制备西红花苷纳米脂质体,粒径(194.86±25.82)nm,包封率(4.6±0.8)%。细胞毒性实验显示,西红花苷对宫颈癌HeLa细胞和乳腺癌MCF-7细胞半数抑制浓度(IC50)分别为1.603、7.35 mmol/L;而西红花苷纳米脂质体IC50则分别为0.61、1.22 mmol/L。表明脂质体显著增强了西红花苷对癌细胞的生长抑制作用。
此外,为延长西红花苷血液滞留时间及增加其被动靶向特性,Rastgoo等[40]对传统纳米脂质体进行聚乙二醇(PEG)修饰,以氢化卵磷脂(HSPC)和Chol为原料,采用溶剂蒸发法制备添加MPEG2000-DSPE的西红花苷脂质体。粒径为(127.6±1.5)nm,包封率为(84.62±0.59)%,多分散指数为0.087±0.018,Zeta电位(−21.7±6.7)mV。体内药理实验表明,与西红花苷相比,西红花苷纳米脂质体显示出更明显的抑制结肠癌生长及提高荷瘤小鼠存活率的药效。另一方面,殷平华[41]研究表明西红花苷脂质体对D-半乳糖胺诱导的急性小鼠肝损伤具有保护作用,推测其作用机制可能是西红花苷嵌入肝脏线粒体膜,清除氧自由基,调节膜结构流动性和稳定性,促使膜上呼吸链功能改善和恢复,以提供维持细胞基本活动所需能量。
2.2 微囊
微囊(microcapsule)是利用高分子材料,将药物包嵌成粒径小于5 000 μm(通常为5~250 μm)微小胶囊,由芯料(药物)及包料(惰性聚合物)组成,广泛应用于提高药物稳定性。西红花酸长共轭结构使其在制备储存中不稳定。Zhou等[42]研究表明,利用阿拉伯树胶(GA)作壁材,通过喷雾干燥法,进气温度210 ℃,液-固比10∶1条件下制备西红花酸微囊外形凹陷、干瘪、表面无裂痕,微囊包埋率为(85.03±1.35)%,并且在低氧条件,半衰期较西红花酸延长4.29倍,稳定性显著提高。Kyriakoudi等[43]也采用相同方法将西红花苷包合于麦芽糖糊精中制备成微囊,结果显示,微囊化增加了西红花苷在胃肠道的稳定性,生物利用度也显著提高(由60%提高至71.9%)。
2.3 复乳
复乳(multiple emulsion)为乳状液分散在其他连续相中形成的多层乳状液,一般有O/W/O和W/O/W 2种类型。在结构上具独特“两膜三相”多隔室结构,如目前广泛研究的W/O/W型复乳,从内到外依次具有内水相、内-水油界面膜、油相、外-油水界面膜、外水相等,属多相体系,故可将性质不同药物分别溶解在不同相中,起到隔离保护、控制释放等作用[44]。Faridi等[45]以西红花提取物、葵花油和司盘80为原料制备W/O型微乳,进一步乳化将其分散在含乳清蛋白浓缩物(WPC)/果胶复合物的外水相中,制备得到西红花苷W/O/W型复乳,包封率达96.66%,表面光滑、无气孔和裂纹。经测试可稳定存放22 d,无分层、沉淀、乳化现象发生。研究显示,西红花苷复乳在胃液释放量为12%,在肠液中释放量高,这是由于果胶聚集体在微碱性溶液中倾向于解离和扩散[46]。提示西红花苷复乳具有控释和缓释特性。
此外,Mehrnia等[47]对传统多重乳液制备工艺进行改进,采用自发乳化法,以非离子表面活性剂司盘80和聚甘油聚蓖麻油酸酯(PGPR)作表面活性剂,制备小粒径西红花苷W/O型纳米乳。随后该课题组采用同样方法制备W1/O型纳米乳,进一步乳化将其分散在含有生物聚合物外水相(W2),并研究Angum gum(AG)、WPC、阿拉伯胶(GA)3种不同聚合物作为西红花苷W/O/W型复乳外水相时,西红花苷稳定性与释放特性[48]。结果显示,由于AG具有高黏性,其制备的乳液最稳定,未出现分层现象。而体外释放实验显示,在肠道环境下,西红花苷释放速率依次为WPC>GA>AG,表明AG具有缓释的作用。
2.4 纳米粒
纳米粒(nanoparticle)是指由天然或合成高分子材料制成的粒径在10~100 nm的固态胶体微粒,包括纳米球和纳米囊。西红花苷纳米粒制备多采用新型高分子材料,通过在其表面使用不同性质材料进行修饰达到缓释、主动靶向等目的。
2.4.1 壳聚糖(Cs)/藻酸钠(Alg)纳米粒 Cs/Alg纳米粒为新型药物载体,可提高药物稳定性,改善药物生物利用度、靶向性、生物黏附性以及赋予药物在胃肠道中的控释特性[49-50]。Rahaiee等[51]采用改进离子凝胶法将西红花苷包载到Cs/Alg纳米粒载体,并研究其稳定性。应用响应曲面法得到载体组成为0.08% Cs和0.10%Alg(pH 4.7);制得的纳米粒粒径为236 nm,ζ电位值、产率、包封率、载药量依次为−33.52 mV、48.33%、38.16%、30.96%;经测试包封后的西红花苷稳定性显著提高。此外,Rahaiee等[52]还以相同方法制备西红花苷Cs/Alg纳米粒并测定其抗癌活性。结果显示,该纳米粒为pH依赖性溶胀,在pH值为1.2的氯化钾-盐酸缓冲液中最高溶胀比为16,在模拟胃液(酸性)环境下,西红花苷可从载体中持续可控释放。此外,与西红花苷相比,西红花苷Cs/Alg纳米粒具有更明显的MCF-7细胞抑制活性,表明包封后的西红花苷抗癌活性增强。
2.4.2 磁性纳米粒 磁性纳米粒(magnetic nanoparticles)为粒径<100 nm磁性纳米材料,具良好生物相容性、生物降解性和超顺磁性等。El-Kharrag等[53]以FeCl2·4H2O、FeCl3和含葡聚糖的NaOH溶液为原料,采用共沉淀法制备磁性纳米粒,再进行原位包合制备西红花酸磁性纳米粒。研究显示,西红花酸磁性纳米粒在水中具良好的分散性和稳定性,呈球形,平均粒径16 nm,相比于西红花酸(3mg/mL),磁性纳米粒仅需0.09mg/mL即可显示相同的HepG2细胞抑制活性,而该西红花苷磁纳米粒在体内是否能在外加磁场条件下靶向到患癌部位发挥疗效,显示高效低毒特性,有待于进一步深入研究。
2.4.3 纳米金 纳米金(goldnanoparticles,AuNPs)粒径<100 nm,具良好生物相容性、性质稳定、易修饰以及无毒等优点,并可提高药物生物活性。Vijayakumar等[54]以HAuCl4·3H2O和西红花提取物为原料,采用一锅法合成粒径为(15±5)nm、稳定均一并具三维立体结构的西红花苷球形纳米金。此外,Hoshyar等[55]还以西红花苷和HAuCl4、NaOH为原料采用相同方法合成粒径为4~10 nm的球形西红花苷-AuNPs。结果显示,西红花苷纳米金可靶向进入MCF-7细胞内并释放出药物,这显示纳米金载体提高了西红花苷对MCF-7细胞靶向性。同时,与西红花苷相比,相同剂量西红花苷纳米金表现出更强的MCF-7细胞抑制活性(P<0.05)。研究还显示,该纳米金可有效保护西红花苷在体内的稳定性,并防止其降解。
2.4.4 乳酸羟基乙酸共聚物 [poly (lactic-co- glycolic acid),PLGA] 纳米粒 PLGA是近年来广泛应用于缓释药物研究的纳米微球载体材料,具有低毒性、良好的生物相容性。通过调整聚合物的相对分子质量、亲水性及乳酸-羟基乙酸比例可控制PLGA降解速度,最终达到缓释目的[56]。Langroodi等[57]采用W/O/W复乳法制备西红花酸PLGA纳米粒。最佳制备工艺为5% PVA,药物与聚合物比例为1∶20,二氯甲烷与丙酮混合液(4∶1)作为有机相,制得纳米粒粒径为178 nm,呈球形且分布均匀;西红花酸的包封率和载药量分别为65%、2.94%;体外释放实验显示,约80%的药物在48 h内逐渐释放,表明西红花酸PLGA纳米粒具有缓释作用。同时,细胞毒性实验显示,与西红花酸相比,西红花酸PLGA纳米粒对MCF-7细胞有更明显的抑制作用。此外,Ghahestani等[58]以PLGA、PVA为原料采用单一乳液-溶剂蒸发法对西红花酸进行包载制备西红花酸PLGA纳米粒。结果显示最佳配方:二氯甲烷为有机溶剂,5% PVA为稳定剂,西红花酸与聚合物比1∶20,制得的纳米粒包封率为97.2%,载药量为4.8%,粒径为288 nm;24 h西红花酸的释放率为(44.49±2.80)%,4 d后药物的释放达到最大值为96.43%,说明PLGA纳米粒可显著延长西红花酸的释放。此外,西红花酸PLGA纳米粒可显著抑制MCF-7细胞生长,其IC50值为(84.73±12.14)μmol/L,而西红花酸为(589.65±5.72)μmol/L,表明包封后的西红花酸抗癌活性明显增强。
2.4.5 PEG功能化硒纳米粒 PEG为pH中性、高水溶性聚合物,具有良好生物相容性。药物经PEG修饰后其半衰期可有效延长,并改善药物动力学和药效等特性,提高靶部位药物浓度。Mary等[59]以Na2SeO3和PEG200为原料制备PEG-SeNPs用来包载西红花苷,制得西红花苷-PEG-SeNPs。结果显示,西红花苷-PEG-SeNPs分散性良好且呈球形,粒径40~50 nm,Zeta电位(−31.360±0.652)mV,载药量8.77%;pH值为5.3时,1 h西红花苷累积释放率为47.0%,48 h为91.0%,pH值为7.4时,1 h累积释放率为11.6%,48 h为34.5%,表明西红花苷在微酸性条件下释放更快,缓释效果更好。因此,PEG-SeNPs可作为癌症治疗的pH介导释放药物递送载体,分布在具有酸性微环境的肿瘤组织周围。研究还显示,与西红花酸相比,西红花苷-PEG- SeNPs显示出更明显的抑制肺癌A549细胞活性。
2.4.6 纳米结构脂质分散体 纳米结构脂质分散体(nanostructured lipid dispersions,NLD)是一种纳米载体,可使药物分子增溶,并保护药物免受降解。为减少西红花酸降解,Esposito等[60]以单油酸甘油酯、胆酸钠和酪蛋白酸钠为原料,制备西红花酸纳米结构脂质分散体。稳定性结果显示,NLD可通过角质层表皮促进西红花酸经皮吸收,并有效抑制西红花酸降解。研究还显示,与西红花酸相比,纳米结构脂质分散体可延长西红花酸抗氧化活性,并提高其抗人黑色素瘤A375细胞活性。
2.5 软胶囊
王震[61]研究制备了西红花总苷卵磷脂复合物自乳化软胶囊。卵磷脂复合物最佳制备工艺:丙酮为反应溶剂,西红花苷与磷脂比1∶1.5,反应1 h,制备的西红花总苷卵磷脂复合物与原料药相比,脂溶性显著增加。卵磷脂复合物与自乳化最佳处方为油酸乙酯-聚山梨酯85-乙醇80.75∶14.25∶5,药物含量8%,制备的西红花总苷卵磷脂复合物自乳化软胶囊在加速实验3个月内稳定。此外,金玉燕[62]制备了西红花多苷自微乳化肠溶胶囊,制备所得胶囊均匀性好,并可显著提高西红花苷生物利用度。
2.6 其他
为提高西红花酸溶解度和生物利用度,钟慧[63]采用溶剂法制备西红花酸固体分散片,最佳处方:西红花酸和聚乙烯吡咯烷酮(PVPK30)比例1∶4。结果显示西红花酸以无定形状态高度分散在载体中,相对生物利用度为原料药的161.18%。此外,为提高西红花酸溶解度,Soltani等[64]将30% PAMAM G4和PPI G4的伯胺烷基化并作为药物递送载体包封西红花酸,此载体可增加药物溶解度和细胞毒性、延长体外释放及提高靶向效率。Selim等[65]研究显示,使用支链淀粉、PVP40和PVP360 3种包封材料分别对西红花苷进行包封可使西红花苷氧化速率显著降低,稳定性提高。
2.7 小结
西红花苷和西红花酸药理活性广泛,但在胃肠道不稳定,同时体内半衰期较短,目前已经研究探索了西红花苷和西红花酸脂质体、微囊、乳剂、纳米粒、软胶囊、缓释片等剂型,利于包合和保护药物,避免药物与胃液、肠液和体液直接接触,减少药物降解,提高药物口服吸收和生物利用度,延长药物体内作用时间。部分剂型还可提高药物稳定性、溶解度和细胞毒性,延长体外释放,使其具缓控释或靶向特性(图3)。经过现代药剂学技术研究后,西红花苷和西红花酸药理功效得到显著增强。

3 结语及展望
西红花苷口服在肠道不稳定,水解成西红花酸,部分西红花酸在小肠黏膜上代谢为西红花酸-单(双)葡萄糖醛酸缀合物,与西红花酸共同进入血液循环分布全身各组织器官,显示广泛药理活性,但其中主要发挥药理功效的成分是西红花酸,还是其单(双)葡萄糖醛酸缀合物,并未见文献报道,还有待于更深入药理学研究,该机制的阐明可有利于西红花苷和西红花酸体内活性成分监测指标的确定。另一方面,有关西红花苷和西红花酸现代递药研究在一定程度上解决了西红花苷和西红花酸溶解性、稳定性差,生物利用度低及体内半衰期短等问题,提高了西红花苷和西红花酸药理活性,但这一系列研究主要采用体外细胞模型,动物体内研究相对欠缺。同时,鉴于西红花苷和西红花酸药理活性广泛,具有明显治疗心血管疾病和中枢神经系统疾病等作用,但以上研究主要侧重西红花苷和西红花酸抗癌活性评价,药效评价不够全面。此外,西红花苷制剂学研究尚缺少毒理学评价,并未见相关临床研究报道,如何利用现代生物学技术对西红花苷现代制剂进行更全面系统研究,将是今后西红花苷和西红花酸的研究方向。
参考文献(略)
来 源:张彩凤,刘银花,王雅溶,汪 泽,陈燕玲,陈 阳. 西红花苷和西红花酸的药动学及制剂学研究进展 [J]. 中草药, 2019, 50(1):234-242.















